

Ano: 1994 Vol. 60 Ed. 4 - Outubro - Dezembro - (9º)
Seção: Relato de Casos
Páginas: 297 a 302
Síndrome de Alport
Alport's Syndrome
Autor(es):
Signe Schuster Grasel*,
Roberta Ribeiro de Almeida**,
Betsy Rayo Aguirre***,
Marco Aurelio Bottino****,
Aroldo Miniti*****.
Palavras-chave: Nefrite hereditária, surdez
Keywords: Hereditary nephritís, deafness
Resumo:
A síndrome de Alport é patologia rara de caráter hereditário cuja descrição clássica envolve a associação entre nefropatia e disacusia neurossensorial simétrica e progressiva. Outras manifestações clinicas associadas menos freqüente são alterações oculares como arco senil, lenticone anterior, catarata e alterações de retina, e também, vestibulopatia periférica.
Os autores apresentam dois casos com sintomatologia cócleovestibular intensa apesar do discreto comprometimento renal. Discutem a necessidade e importância da abordagem diagnóstica e terapêutica dos sintomas vestibulares, uma vez que estes podem ser a principal queixa do paciente.
Abstract:
Alport's syndrome is a hereditary disease characterized by the association of nephropathy with symmetrical and progressive sensorineural hearing loss. Other clinical manifestations Ne ocular arcus senilis, anterior lenticonus, cataratt, retinal changes, and peripheral vestibular lesion are less common.
The authors present two patients with substantial auditory and vestibular symptams despite little renal involvement. Since the vestibular disorder may be the patient's main complaint the importante of this appropriate diagnosis and management is diseussed.
![]()
INTRODUÇÃO
A síndrome de Alport é uma nefropatía hereditária associada à dísacusia neurossensorial bilateral progressiva, descrita pela primeira vez em 1927 por Alport. Foi Alport que descobriu a alta incidência de perda auditiva neurossensorial em membros de uma família com nefrite hereditária. Quatro gerações da família tinham sido estudadas por vários autores em um período de 25 anos. Alport concluiu que ambas as patologias faziam parte de uma síndrome. A via de transmissão genética, ainda controvertida, é descrita como através de gene dominante ligado ao cromossomo X 1. A observação clínica que o quadro é mais grave em homens que em mulheres favorece esta teoria, determinada por expressão completa do gene no sexo masculino e incompleta no sexo feminino (hipótese de Lyons).
Outras teorias incluem transmissão autossômica dominante com seleção do gene defeituoso associado a um cromossomo X2, pleiotropia e penetração incompleta e heterogenia 3
Acredita-se que uma alteração no metabolismo de dois glicosídeos resulta em produção anómala do colágeno da membrana basal.
Estudos histopatológicos realizados após biópisia renal mostram as mais variadas alterações: espessamento da parede capilar em glomerulos dispersos, áreas localizadas de solidificação em glomerulos, células espumosas (foam cells) no córtex renal, atrofia tubular de grau variável, podendo chegar ao quadro de glomerulonefrite focal proliferativa na doença progressiva.
A microscopia eletrônica é de grande valor, mostrando espessamento e desintegração da membrana basal dos glomerulos, patognomónico para esta patologia 4.
Os autores concordam que a característica das lesões renais é supostamente a combinação de alterações de glomerulonefrite crônica, pielonefrite e nefrite intersticiais 5,6.
Os ossos temporais de pacientes com Alport mostram alterações principalmente cocleares. As mais freqüentes são atrofia da estria vascular, vacuolização do ligamento espiral e degeneração de células ciliadas e ganglionares 7.
Permanece a dúvida quais das alterações sejam artefatos de autólise e quais devido à patologia de bases.
As manifestações clínicas envolvem sintomas renais como proteinuria, hematuria e hipertensão arterial podendo evoluir para a insuficiência renal crônica principalmente no sexo masculino.
A disacusia neurossensorial bilateral, simétrica, progressiva é associada em certos casos à vestibulopatia periférica com hiporreflexia na prova ca16rica.
Cerca de 15% dos pacientes manifestam alterações oculares. Arco senil, lenticone, catarata e alterações de retina são os achados mais encontrados pouco interferindo, porém, na acuidade visual dos pacientes.
O objetivo deste trabalho é apresentar dois doentes com síndrome de Alport, enfatizando os sintomas vestibulares associados, uma vez que estes podem interferir sensivelmente na qualidade de vida dos pacientes.
CASUÍSTICA
Trata -se de duas irmãs , filhas de pais consangüíneos naturais do interior de Alagoas, com história familiar de hipertensão arterial sistêmica, hipoacusia e diabetes melitus. Os pais faleceram ambos de acidente vascular cerebral.
Caso 1:
M. R. S., 37 anos, refere hipoacusia progressiva bilateral desde os 7 anos de idade acompanhada de zumbido tipo estalido e diminuição de acuidade visual há 6 anos. Ela apresenta vertigem tipo desequilíbrio em crises acompanhado de sintomas neurovegetativos e piora da hipoacusia e do zumbido. Há 4 anos foi feito o diagnóstico da nefropatia e hipertensão arterial sistêmica (HAS).
Os exames laboratoriais mostram: glicemia 154 mg%, uréia 27 mg / 100 ml, creatinina 1,4 mg / 100 ml, Na 135 meq/ ml, K 4,2 meq/ml: Urina I com proteinuria de 2,15 g 1.
A biópisia renal foi sugestiva de nefroesclerose benigna.
Da avaliação oftalmológica consta lenticone anterior.
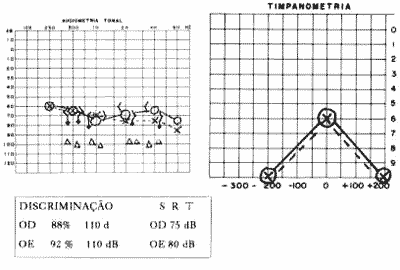
O exame de ORL completo é normal. A audiometria revela disacusia neurossensorial bilateral simétrica com curva plana e limiar em 60 dB, discriminação em 88% OD e 92% OE à 110 dB e presença de recrutamento. As alterações são sugestivas de disacusia coclear (Fig. 1).
As alterações encontradas no exame otoneurológico são: nistagmo posicional para a direita em posição de Rose, da prova calórica resulta preponderância direcional de 36% para direita e predomínio labiríntico de 16%, apontando um quadro de vestibulopatia periférica.
Diagnóstico: nefropatia, HAS, diabetes melitus, lenticone anterior e cocleovestibulopatia periférica - síndrome de Alport.
Caso 2:
M. R. S., 34 anos é a irmã mais nova de M. R. S. (37 anos). Ela refere também hipoacusia progressiva desde os 8 anos de idade e vertigem rotatória objetiva em crises, acompanhada de zumbido agudo bilateral há 17 anos. Ela usa prótese auditiva no OD. Apresenta ainda diminuição da acuidade visual há 3 anos. Normotensa, ela é acompanhada no serviço de Nefrologia após o achado de hematuria microscópica em exame de rotina. Ela recusou a realização de biópsia renal.
Os exames laboratoriais revelam: glicemia de 124 mg%, uréia 28 mg 1100 ml, creatinina 0,8 mg/ 100 ml, Na 135 meq / ml, K 4 meq / ml e urina 1 com proteínas 0,3 g / 1, 2 leucócitos / campo, 5 eritrócitos / campo.
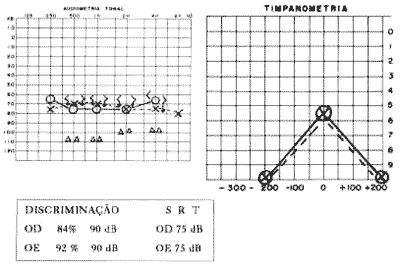
Com exame ORL normal, apresenta na audiometria disacusia neurossensorial bilateral simétrica com limiar de 75 dB, discriminação de 84% OD e 92% OE à 90 dB e SRT compatível em 75 dB, sugestivo de cocleopatia (Fig. 2).
No exame otoneurológico encontramos as seguintes alterações: nistagmo espontâneo horizontal à direita de olhos abertos, nistagmo posicional para a direita nos decúbitos laterais, em posição de Rose e sentado, e na prova calórica hiporreflexia (4D / s) à 30' no OD resultando em preponderância direcional de 33% para a direita e predomínio labiríntico de 16% para a esquerda, sugestivo de vestibulopatia periférica deficitária à direita.
Diagnóstico: nefropatia, glicemia limitrofe, cocleovestibulopatia periférica - síndrome de Alport.
Figura 1 - Audiometria e impedânciometria de M. R. S., 37 anos (caso 1).
Figura 2 - Audiometria e impedânciometria de M. R. S., 34 anos (caso 2).
DISCUSSÃO
A associação de nefropatia com disacusia neurossensorial progressiva sempre levantou dúvidas se a disacusia não era apenas conseqüência das alterações renais. A interferência de drogas ototóxicas, insuficiência renal crônica, hemodiálise, transplante renal e hipertensão arterial não pode ser subestimada, uma vez que muitos pacientes fazem audiometria apenas no estado avançado da doença. Em pacientes com insuficiência renal crônica (IRC) Oda et al 8 demonstraram perda auditiva proporcional ao número de sessões de hemodiálise. A influência do transplante renal sobre a audição em pacientes com síndrome de Alport merece estudos mais aprofundados. Nenhum caso descrito teve melhora da audição após o transplante, contrastando com as melhoras auditivas documentadas em doentes com outras causas de IRC'. Estabilização de audição em casos isolados 10 ou mais comum, perda progressiva após o transplante, levam a dúvidas quanto ao benefício do transplante para o nível de audição em doentes com síndrome de Alport. O distúrbio hidroeletrolítico, principalmente a hiponatremia, já foi responsabilizada pela perda auditiva. Existem evidências correlacionando o grau de hiponatremia com o grau de perda auditiva, perda que pode ser parcialmente reversível após correção dos níveis séricos de sódio 11. Controvertido é também o efeito da hipertensão arterial sobre o grau de perda auditiva.
As nossas pacientes apresentam normanatremia e função renal compensada. Nenhuma delas tem história de uso de ototóxicos, não foi submetida à transplante renal nem à hemodiálise. O grau de hipertensão arterial também não parece ter influenciado o nível de audição de nossas pacientes. A paciente (Caso 1) com HAS documentada tem limiar tonal melhor (60 dB) que a irmã (Caso 2) normotensa (75 dB).
Concluímos que em nossas pacientes a função renal pouco interferiu na progressão da perda auditiva.
A história familiar nem sempre ajuda no diagnóstico. Como já mencionou Gleeson 4, ela pode ser negativa para surdez como em nossas pacientes. A consangüinidade dos pais poderia sugerir outra causa para a surdez das filhas, uma vez que consangüinidade figura ainda entre as principais etiologias de deficiência auditiva congênita em nosso meio 12.
Pouco se fala sobre afecções vestibulares em pacientes com síndrome de Alport. Alvares Cruz e col. 10 submeteram três pacientes, portadores da síndrome, ao exame otoneurológico e não encontraram alterações. Miller e col. 5 estudaram cinco pacientes. Os quatro submetidos ao exame otoneurológico apresentaram hiporreflexia. O último paciente faleceu antes de poder realizar o exame. O estudo dos seus ossos temporais é um dos poucos que nos dá informações sobre alterações histopatológicas do aparelho vestibular. Míllers descreveu formações císticas embaixo da mácula do Báculo, alterações parecidas às encontradas por Winter e col 13 no ligamento espiral da mácula do utrículo. Fujita e Hayden 14 observaram que o achado histopatológico principal era a ausência de alterações patológicas consistentes ao nível do ouvido interno, mas encontraram a camada granular acentuada entre a epitélio neurossensorial e a membrana otolítica da mácula do Báculo assim como glóbulos na superfície do órgão de Corti.
As nossas pacientes referem queixas de vertigem que no decorrer dos anos se tornaram o motivo principal das visitas ao hospital, dado o discreto comprometimento renal e a estabilização dos parâmetros metabólicos. No exame otoneurológico ambas apresentam sinais de vestibulopatia periférica com preponderância direcional significante, uma delas (Caso 2) com hiporreflexia importante (4') na prova calórica à 30° no ouvido direito.
Queremos ressaltar a importância da investigação diagnóstica e do tratamento dos sintomas vertiginosos nos portadores da síndrome de Alport.
Além da disfunção cócleovestibular muitos pacientes apresentam também um comprometimento visual, resultando em déficit funcional de dois sistemas sensitivos intimamente relacionados à manutenção do equilíbrio.. Com maior dificuldade de compensar distúrbios de equilíbrio estes pacientes se tornam muitas vezes inseguros e ansiosos. A colocação de prótese auditiva conforme o grau de disacusia, o acompanhamento das patologias renais, metabólicas e da HAS devem ser complementados por um acompanhamento periódico com um otoneurologista para garantir aos pacientes com vestibulopatia o tratamento adequado dos sintomas de vertigem e da ansiedade concomitante.
REFERÊNCIA BIBLIOGRÁFICA
1. O'NEILL, V. N.; ATKIN, C. L.; BLOOMER, H. A. - Hereditary nephritis: a reexamination of its clinical and genetic features. Ann Intern Med 88: 176, 1978.
2. COHEN, M. M.; CASSIDY, G.; HANNA, B. L. - A genetic study of hereditary renal dysfunction with associated nerve deafness. Am J Hum Genet 13: 379 - 389, 1961.
3. GRAHAM, J. B. - Hereditary chronic kidney disease: an alternative to partial sex-linkage in the Utah kindred. Am J Hum Genet 11: 333 - 338, 1959.
4. GLESSON, M. J. - Alport's synsdrome: audiologieal manifestations and implications. J Laryngol Otol 98: 449 - 465, 1984.
5. MILLER, G. W.; JOEPH, D. J.; COZAD, R. L.; McCABE, B. F. - Alport's syndrome. Arch Otolaryngol92: 419 - 432, 1970
6. TURNER, J. S. -Hereditary hearing Iosswith nephropathy (Alport'syndrome). Acta Otolaryngol Supp1271: 8 - 26, 1970.
7. JOHNSSON, L. G.; ARENBERG, K. - Cochlear abnormalities in Alport's syndrome. Arch Otolaryngol 107: 340 - 349, 1981.
8. ODA, M.; PRECIADO, M.; QUICK, C.; PAPARELLA, M. - Labyrithine pathology of chronic renal failure patients treated with haemodialysis and kidney transplantation. Laryngoscope 84: 1489 - 1506, 1974.
9. McDONNALD, J, J.; ZINCKE, H.; ANDERSON, C. F. - Hearing loss improvement with transplantation. Laryngoscope 88: 38 - 42, 1978.
10. ALVARES CRUZ FILHO, N.; CUBAS DE ALMEIDA, S. I.; OTTO, P.; FORMIGONI, L. G.; MINITI, A.; SARAIVA MOREIRA, R. - Le syndrome d'Alport: étude oto-neurologique et génétique de deux familles. Rev. Larvngol 104: 449 - 452, 1983.
11. YASSIN, A.; BADRY, A.; FATT-HI, A. - The relationship between electrolyte balance and cochlear disturbances in cases of renal failure. J Laryngol Otol 84: 429 - 435, 1976.
12. MEDICIS DA SILVEIRA, J. A. - Estudo da deficiência auditiva em crianças submetidas a exames de potenciais evocados auditivos. São Paulo, 1992. p. 40 Tese de Doutoramento apresentado à Faculdade de Medicina da Universidade de São Paulo.
13. WINTER, L. E.; ORAM, B. M.; BANOWETZ, J. D. - Hearing loss in hereditary renal disease. Arch Otolaryngol 88: 238 - 241, 1968.
14. FUJITA, S.; HAYDEN, R. C. - Alport's syndrome. Arch Otolaryngo190: 453 - 466, 1969.
* Pós-graduanda da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP.
** Médica residente da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP.
*** Médica ex-residente da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP.
**** Médico assistente da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP.
***** Professor titular da Disciplina de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP.
Este trabalho foi realizado na Clínica de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da USP e no LIM-32 (Serviço do Prof. Dr. Aroldo Miniti)
Endereço para correspondência: Av. Dr. Enéas de Carvalho Aguiar, 255 - 6° andar - São Paulo - SP - CEP 05403-000.
Este trabalho foi apresentado na Sessão de Tema Livre do XXXI Congresso Brasileiro de Otorrinolaringologia em São Paulo, 15 a 20 de agosto de 1992.
Artigo recebido em 9 de maio de 1994.
Artigo aceito em 20 de maio de 1994.
Imprimir: ![]()