

Ano: 1998 Vol. 64 Ed. 1 - Janeiro - Fevereiro - (9º)
Seção: Relato de Casos
Páginas: 68 a 71
Síndrome de Fraser e suas Manifestações otorrinolaringológicas. Relato de um caso.
The Fraser Syndrome and its Otorhinolaryngeal Manifestatios: A case report.
Autor(es):
Marcos Moceliin* ;
Maurício Buschle** ;
Carla Rogenski*** ;
Odilon F Neto **** ;
Luciana Gabardo Stahlke*****;
Mirian B. O. Kôning*****.
Palavras-chave: síndrome de Fraser, criptoftalmia
Keywords: Fraser syndrome, cryptophtalmia
Resumo:
A síndrome de Fraser é uma rara desordem cromossômica autossômica recessiva, descrita pela primeira vez em 1962 (Fraser), cuja característica clínica mais consistente é a criptoftalmia, entre outras manifestações oftálmicas, associada a várias anomalias crânio-facial e sistêmicas, incluindo malformações de ouvido (pavilhão auricular, conduto auditivo externo, ouvido médio e ouvido interno), malformações do nariz e da laringe; bem como alterações urogenitais, gastrintestinais, osteo-esqueléticas e do sistema nervoso central. O diagnóstico é feito por ocasião do nascimento, frente às óbvias alterações, e, algumas vezes, na ultrassonografia pré-natal. O tratamento é dependente das malformações urogenitais e laríngeas associadas. Apresentamos um caso que vem sendo acompanhado em nossa instituição há seis anos, apresentando diversas características da síndrome, com ênfase às alterações otorrinolaringológicas, em contribuição à pequena quantidade de informações específicas em ORL existente na literatura.INTRODUÇÃO
A síndrome de Fraser é rara desordem cromossômica autossômica recessiva e, por isso, sua expressão fenotípica depende da consangüinidade dos pais. Descrita pela primeira vez por Fraser, em 1962, já havia relato de criptoftalmia desde 1872 (Zehender), o qual observou a continuidade do epitélio dos olhos com a face. François, em 1965, descreveu quatro das características da síndrome: malformações de ouvido e nariz, sindactilia e alterações da genitália.
Grande número de anormalidades fazem parte da síndrome de Fraser, e foram reportadas por Gupta:
Otológicas.
Estenose ou atresia do canal auditivo externo,
baixa implantação de orelha,
defeitos do pavilhão e
orelha fendida.
Nasais.
Coloboma de asa nasal,
atresia de coanas,
base nasal alargada,
narinas hipoplásicas e estreitadas e
chanfradura nas narinas.
Laríngea.
Atresia ou estenose.
Oftalmológicas.
Criptoftalmia (parcial ou completa),
simbléfaro congênito,
hipertelorismo,
microftalmia,
coloboma de pálpebra superior e
sobrancelha supranumeráría.
Urogenitais.
Agenesia renal,
hipospádia,
criptorquidia,
hipertrofia de clitóris,
útero bicomo,
fusão de pequenos lábios e
malformações das trompas.
Gerais.
Fendas labial e palatina,
sindactilia, hérnia umbilical,
meningoencéfalocele, retardo mental,
estenose ou atresia anal,
má fusão da sínfise púbica,
hipertelorismo mamário,
implantação dos cabelos próximo à porção lateral da região frontal,
mama supranumerária e fácies de Potter.
O diagnóstico é feito por ocasião do nascimento, frente às óbvias alterações, e, algumas vezes, à ultra-sonografia pré-natal e fetoscopia. O tratamento é dependente das malformações presentes e O prognóstico relaciona-se à gravidade das malformações urogenitais e laríngeas. Apresentamos um caso, o qual vem sendo acompanhado em nossa instituição há oito anos, apresentando diversas características da síndrome, com ênfase às alterações otorrinolaringológicas (investigadas através de exame ORL, métodos de imagem, tais como tomografia computadorizada de ouvidos, mastóide, nariz e seios paranasais, assim como a nasofibrófaríngoscopia para as alterações das cavidades nasais, rinofaringe e tuba auditiva, orofaringe, hipofaringe e laringe). Procuramos a correção das anomalias anatômicas, tão precocemente quanto possível e seguro, para serem evitadas as complicações decorrentes das complexas alterações pertinentes à síndrome.
RELATO DE CASO
L . A. A. F., com nove anos de idade, filha de pais consangüíneos, natural de Curitiba, PR.
Paciente com história de otorréia purulenta e fétida há quatro meses em ouvido esquerdo, acompanhada de hipoacusia.
Com um ano e um mês de idade, foi encaminhada ao Departamento de Genética para avaliação: concluiu-se que se tratava de síndrome dismórfica de expressividade variável, ocasionada por gene autossômico recessivo, resultante da consangüinidade dos pais, primos em primeiro grau. Foi realizado estudo cromossômico, sendo a constituição cromossômica normal 46XX. O aconselhamento genético demonstrou 25% de possibilidade em relação à manifestação da síndrome.
Como antecedentes patológicos, temos a história de obstipação intestinal; com um ano e dois meses, foi submetida a cirurgia para correção de simbléfaro pela oftalmologia, sendo que, três meses depois, as pálpebras soldaram-se novamente. Possui ecografia cerebral normal, eletroencefalograma normal e exame ortopédico normal.
EXAME OTORRINOLARINGOLÓGICO
boca
Palato ogival.
Nariz
Pirâmide nasal deprimida,
nariz bífido,
fendas oblíquas,
base nasal alargada e achatada,
com sulcos horizontais,
ápice nasal hipoplasico,
narinas separadas e anteriorizadas e
duplo sulco nasolabial mediano.
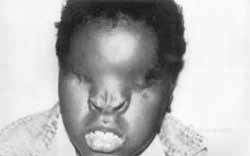 Figura 1. Paciente com 9 anos apresentando pirâmide nasal deprimida, base nasal alargada, narinas separadas e anteriorizadas, nariz bífido e fendas oblíquas.
Figura 1. Paciente com 9 anos apresentando pirâmide nasal deprimida, base nasal alargada, narinas separadas e anteriorizadas, nariz bífido e fendas oblíquas.
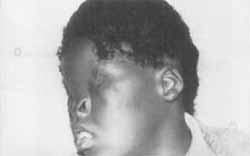 Figura 2. Pode se verificar leve microtia, hélice dobrada, ausência de sobrancelhas e cílios, implantação do cabelo próximo à região ocular e ausência de fissura palpebral.
Figura 2. Pode se verificar leve microtia, hélice dobrada, ausência de sobrancelhas e cílios, implantação do cabelo próximo à região ocular e ausência de fissura palpebral.
* Professor Titular e Chefe da Disciplina de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
** Professor Assistente da Disciplina de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
*** Médica Especializando em Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná. Médico Residente em Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
**** Acadêmica do 6º Ano de Medicina do Hospital de Clínicas da Universidade Federal do Paraná.
***** Médica Otorrinolaringologista.
O presente trabalho está vinculado ao Departamento de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná. Endereço para correspondência: Luciana Gabardo Stahlke - Rua Bispo Dom José, 2788 - Seminário - Curitiba /PR - CEP 80440-080 - Telefone: (041) 242-8835. Apresentações: X Jornada do HC - UFPR, em 4 de outubro de 1996, em Curitiba /PR, XXXIII Congresso Brasileiro de Otorrinolaringologia, em 4 de novembro de 1996, em Recife /PE. Artigo recebido em 11 de junho de 1997. Artigo aceito em 1 de outubro de 1997.
Abstract:
The Fraser syndrome, described for the first time in 1962 (Fraser), is a rare chromosomic autosomal recessive disorder, being its most consistent clinical characteristic the cryptophtalmia, among other ophtalmological manifestations, associated with various cranio-facial and systemic abnormalities, including malformations of the ear (auricle,external auditory canal, middle ear or internal ear), malformations of the nose and the larynx, as well as urogenital, gastrintestinal, osteo-skeletal and central nervous system alterations. The diagnosis is made at birth, in the presence of obvious alterations, and sometimes during prenatal ultrasound scanning. The treatment is contingent on the existing malformations, and the prognosis is related to the seriousness of the associated urogenital and laryngeal malformations. As a contributions to the scarce specific informations about ORL, existing in literature we present a case which has been accompanied in our institution for the last six years, showing various characteristics of the syndrome and emphasizing the otolaryngeal alterations.
![]()
INTRODUÇÃO
A síndrome de Fraser é rara desordem cromossômica autossômica recessiva e, por isso, sua expressão fenotípica depende da consangüinidade dos pais. Descrita pela primeira vez por Fraser, em 1962, já havia relato de criptoftalmia desde 1872 (Zehender), o qual observou a continuidade do epitélio dos olhos com a face. François, em 1965, descreveu quatro das características da síndrome: malformações de ouvido e nariz, sindactilia e alterações da genitália.
Grande número de anormalidades fazem parte da síndrome de Fraser, e foram reportadas por Gupta:
Otológicas.
Estenose ou atresia do canal auditivo externo,
baixa implantação de orelha,
defeitos do pavilhão e
orelha fendida.
Nasais.
Coloboma de asa nasal,
atresia de coanas,
base nasal alargada,
narinas hipoplásicas e estreitadas e
chanfradura nas narinas.
Laríngea.
Atresia ou estenose.
Oftalmológicas.
Criptoftalmia (parcial ou completa),
simbléfaro congênito,
hipertelorismo,
microftalmia,
coloboma de pálpebra superior e
sobrancelha supranumeráría.
Urogenitais.
Agenesia renal,
hipospádia,
criptorquidia,
hipertrofia de clitóris,
útero bicomo,
fusão de pequenos lábios e
malformações das trompas.
Gerais.
Fendas labial e palatina,
sindactilia, hérnia umbilical,
meningoencéfalocele, retardo mental,
estenose ou atresia anal,
má fusão da sínfise púbica,
hipertelorismo mamário,
implantação dos cabelos próximo à porção lateral da região frontal,
mama supranumerária e fácies de Potter.
O diagnóstico é feito por ocasião do nascimento, frente às óbvias alterações, e, algumas vezes, à ultra-sonografia pré-natal e fetoscopia. O tratamento é dependente das malformações presentes e O prognóstico relaciona-se à gravidade das malformações urogenitais e laríngeas. Apresentamos um caso, o qual vem sendo acompanhado em nossa instituição há oito anos, apresentando diversas características da síndrome, com ênfase às alterações otorrinolaringológicas (investigadas através de exame ORL, métodos de imagem, tais como tomografia computadorizada de ouvidos, mastóide, nariz e seios paranasais, assim como a nasofibrófaríngoscopia para as alterações das cavidades nasais, rinofaringe e tuba auditiva, orofaringe, hipofaringe e laringe). Procuramos a correção das anomalias anatômicas, tão precocemente quanto possível e seguro, para serem evitadas as complicações decorrentes das complexas alterações pertinentes à síndrome.
RELATO DE CASO
L . A. A. F., com nove anos de idade, filha de pais consangüíneos, natural de Curitiba, PR.
Paciente com história de otorréia purulenta e fétida há quatro meses em ouvido esquerdo, acompanhada de hipoacusia.
Com um ano e um mês de idade, foi encaminhada ao Departamento de Genética para avaliação: concluiu-se que se tratava de síndrome dismórfica de expressividade variável, ocasionada por gene autossômico recessivo, resultante da consangüinidade dos pais, primos em primeiro grau. Foi realizado estudo cromossômico, sendo a constituição cromossômica normal 46XX. O aconselhamento genético demonstrou 25% de possibilidade em relação à manifestação da síndrome.
Como antecedentes patológicos, temos a história de obstipação intestinal; com um ano e dois meses, foi submetida a cirurgia para correção de simbléfaro pela oftalmologia, sendo que, três meses depois, as pálpebras soldaram-se novamente. Possui ecografia cerebral normal, eletroencefalograma normal e exame ortopédico normal.
EXAME OTORRINOLARINGOLÓGICO
boca
Palato ogival.
Nariz
Pirâmide nasal deprimida,
nariz bífido,
fendas oblíquas,
base nasal alargada e achatada,
com sulcos horizontais,
ápice nasal hipoplasico,
narinas separadas e anteriorizadas e
duplo sulco nasolabial mediano.
Figura 1. Paciente com 9 anos apresentando pirâmide nasal deprimida, base nasal alargada, narinas separadas e anteriorizadas, nariz bífido e fendas oblíquas.
Figura 2. Pode se verificar leve microtia, hélice dobrada, ausência de sobrancelhas e cílios, implantação do cabelo próximo à região ocular e ausência de fissura palpebral.
Ouvido
Leve microtia,
hélice dobrada e lobos anteriorizados,
estenose de conduto auditivo externo, à esquerda, presença de pólipo em ouvido esquerdo,
acompanhado de otorréia amarela e fétida.
Face
Ausência de fissura palpebral,
ausência de sobrancelhas e cílios e malformação do dueto nasolacrimal.
Gerais
Mamilo extranumerário e
atraso no desenvolvimento da genitália.
EXAMES COMPLEMENTARES
Tomografia computadorizada de ouvido e mastóide: hipopneumatização de ambas as mastóides, com área de erosão óssea, preenchidas por tecido mole, sugestivas de colesteatoma.
Tomografia computadorizada de nariz e seios da face: malformação do dueto nasolacrimal à esquerda. Nasofibrobroncoscopia: normal.
Tomografia computadorizada da laringe: normal.
COMENTÁRIOS
Há, na atualidade, mais de cem casos de síndrome de Fraser relatados: a maioria pela oftalmologia e pela genética. Há menor número de casos descritos pela Otorrinolaringologia. Trata-se de síndrome disformica autossômica recessiva. Para seu diagnóstico foram adotados os critérios de Thomas et al., 1986, que se baseiam em critérios maiores e menores. Entre os maiores estão: criptoftalmia, sindactilia e anormalidades da genitália. Entre os menores estão: malformações de ouvido, nariz e laringe, fenda labial ou palatina, defeitos do esqueleto, hérnia umbilical, agenesia renal e retardo mental. O diagnóstico é feito através de 2 critérios maiores e um menor ou um critério maior e quatro menores.
Segundo a revisão de 68 casos feita por Gattuso, as anormalidades craniofaciais da síndrome de Fraser são encontradas nas seguintes proporções: a criptoftalmia aparece em 93% dos casos, anomalias de ouvido, em 44x/0, anomalias em nariz, em 37%, malformações do sistema urinário, em 37%, sistema genital, em 49% e a sindactilia está presente em 54 % dos casos.
O caso descrito em nosso serviço apresenta-se dentro dos parâmetros diagnósticos e da literatura, pois a paciente possui criptoftalmia, atraso no desenvolvimento da genitália e malformações de ouvido e nariz.
Na literatura, existem relatos (Shauer7) em que o diagnóstico pode ser feito no período pré-natal, através de ultrassonografia e fetoscopia.
O prognóstico das pessoas afetadas por essa síndrome depende da intensidade das malformações renais e do aparelho respiratório.
As propostas de tratamento para o caso relatado foram mastoidectomia radical para o colesteatoma à esquerda, tubo de ventilação de longa permanência no ouvido direito e cirurgia estética para correção das deformidades nasais.
REFERÊNCIAS BIBLIOGRÁFICAS
1. FRASER, C. R. - Our genetical load: A review of some aspects of genetical variations. Ann. Hum.. Gener, 25: 387415, 1982.
2. FRANCOIS, J. - Syndrome malformaty avec cryptophthalmies. Ophthalmologica, 150: 215-218, 1965.
3. GUPTA, S. P.; SAXENA, R. C. - Cryptophthalmos. British Journal of Ophthalmology, 46 629-632, 1962.
4. THOMAS, 1. T.; FRIAS, J. L.; FELIX, V.; SANCHES, L. L.; HERNANDES, R. A.; JONES, M. C. - Isolated and syndrommc cryptophthalmos. American journal of Medical Gnetics, 25: 85-88, 1986.
5. GATTUSO, J.; PATTON, M. A.; BARAITSER, M. - The clinical spectrum of the Fraser syndrom: report of three new cases and review. Journal of Medical Genetics, 24: 549-555, 1987.
6. SCHAUER, G. M.; DUNN, L. K.; GODMILOW, L.; EAGLE, R. C. JR. et al. - Prenatal diagnosis of Fraser syndrome at 18,5 weeks gestation, with autopsy findings at 19 weeks. Am Journal ofMedical Genetics, 37 583-591, 1990
7. MINA, M. M. F.; GRENBERG, C.: LEVIN, B. - ENT abnormalities associated with Fraser syndrom: case report and literature review. Tbe journal of Otolaryngology, 17 (5): 233-36, 1988.
8. FORD, G. R.; IRVING, R. M.; JONES, N. S. et al. - ENT manifestations of Fraser syndrome. bejournal ojlaryngology and Otology. London, 106, 1 - 4, 1992.
* Professor Titular e Chefe da Disciplina de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
** Professor Assistente da Disciplina de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
*** Médica Especializando em Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná. Médico Residente em Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná.
**** Acadêmica do 6º Ano de Medicina do Hospital de Clínicas da Universidade Federal do Paraná.
***** Médica Otorrinolaringologista.
O presente trabalho está vinculado ao Departamento de Otorrinolaringologia do Hospital de Clínicas da Universidade Federal do Paraná. Endereço para correspondência: Luciana Gabardo Stahlke - Rua Bispo Dom José, 2788 - Seminário - Curitiba /PR - CEP 80440-080 - Telefone: (041) 242-8835. Apresentações: X Jornada do HC - UFPR, em 4 de outubro de 1996, em Curitiba /PR, XXXIII Congresso Brasileiro de Otorrinolaringologia, em 4 de novembro de 1996, em Recife /PE. Artigo recebido em 11 de junho de 1997. Artigo aceito em 1 de outubro de 1997.
Imprimir: ![]()