

Ano: 1994 Vol. 60 Ed. 1 - Janeiro - Março - (15º)
Seção: Relato de Casos
Páginas: 69 a 72
Síndrome de Alport
Alport Sindrome
Autor(es):
Paulo H. A. Morales,
Mirian S. Peixeira,
Clóvis A. L. Pinto,
Edmir A. Lourenço,
Clemente I.R. Almeida
Palavras-chave: nefrite familiar, surdez
Keywords: Hereditary nephritis, deafness
Resumo:
A Síndrome de Alport é uma doença de caráter progressivo e hereditário, caracterizada pela tdade: nefrite crônica, disacusia neuro-sensorial e lenticone anterior.
Apresentamos o relato de um caso, enfatizando seu diagnóstico clínico.
Abstract:
The Alport syndrome is a disease with progressive and hereditary caracteristics, figured by the triad. cronic nephritis, sensorineural hipoacusia, lenticona anterior.
We show the case report with its evolution in three years period, emphaticising its clinical diagnostic.
![]()
INTRODUÇÃO
A Síndrome de Alport foi descrita pela primeira vez pelo autor de mesmo nome em 1927, quando este passou a relacionar quadros de nefrite crônica hereditária com hipoacusia'. Sohar (1954) observou alterações visuais acompanhando a síndrome, mas apenas em 1959, Nieth descreveu o lenticone anterior como sendo a alteração oftalmológica mais freqüente e atualmente considerada patognomônica por muitos autores. Hoje esta síndrome é caracterizada pela tríade clássica de nefrite crônica hereditária, disacusia neurosensorial progressiva e lentecone anterior.
A hematúria é o sintoma inicial mais freqüente da doença, podendo ser macroscópica ou não, presente desde o nascimento ou nos primeiros meses de vida. A hematúria macroscópica no adulto é rara, sendo nessa fase comum a detecção de hematúria microscópica, cilindros hemáticos e proteimíria ao exame de urina I. Há evolução progressiva para insuficiência renal crônica, chegando ao estágio terminal e óbito porvolta da terceira década devida, com pior prognóstico para indivíduos do sexo masculino e que apresentam proteinúria. As mulheres raramente apresentam insuficiência renal, mas têm a tendência a desenvolverem hipertensão arterial e edema na gravidez;. A biópsia renal ainda é considerada por muitos autores como imprescindível ao diagnóstico da síndrome de Alport, mas as alterações histológicas à microscopia óptica são específicas apenas na fase terminal da insuficiência renal. Outros autores relatam alterações precoces altamente sugestivas à microscopia eletrônica, como mudança na espessura, contorno e densidade eletrônica da membrana glomerular, representadas por lamelação da lâmina densa da membrana glomerular basal, mas mesmo estas não são consideradas patognomônicasz,9.
A disacusia de 20 dB ou mais é um dos achados clássicos, sendo geralmente bilateral e nem sempre clinicamente evidente, com tendência a piorar na adolescência. Perkoff (1951) descreveram dois tipos de curva audiométricas: tipo descendente, mais acentuada em agudos, e curva com queda em 4000 Hz. Considera-se que a lesão é ao nível das células ciliadas do órgão de Corti, afetadas talvez por acúmulo de substâncias tóxicas nos líquidos do labirinto, resultante das alterações metabólicas e bioquímicas, o que explicaria o aparecimento tardio da hipoacusia e sua evolução lenta e progressiva; apesar de ainda não ter sido detectada nenhuma substância tóxica nos líquidos do labirinto. A porção vestibular fica intacta provavelmente porque seu desenvolvímento embrionário ocorre antes do coclear, não dando tempo das toxinas lesá-lo, ou pela presença de um geri com efeito específico sobre o órgão de Corti. As provas calóricas são normais e os achados audiométricos que falam a favor de cocleopatia são: presença de recrutamento, altas porcentagens na prova de SISI, baixas porcentagens na logoaudiometria e curva tipo II na audiometria automática de Békesy'.
Das alterações oftalmológicas, o lenticone anterior é o achado mais freqüente e, de maneira geral, é facilmente visível à biomicroscopia. No entanto, algumas vezes esse abaulamento pode ser muito discreto, apenas observado com uma ampla midríase e com o auxílio do uso do reflexo vermelho através da biomicroscopia direta. Os achados de microscopia óptica revelaram uma cápsula anterior afinada com diminuição de célulasepiteliaisa.
Já foi descrita a presença de lenticone posterior associada a síndrome de Alport, porém sempre acompanhado de lenticone anterior. Outros achados também descritos em literatura são: distrofia polimorfa posterior, maculopatia, arco comeano, cataratas subcapsulares anteriores e ruptura de cápsula anterior do cristalino'. Não há alterações de pressão ocular, na gonioscopia ou fisiologia do humor aquoso.
Otipo de transmissão genética é ainda bastante controverso, mas a teoria mais aceita é a de transmissão através de um geri autossômico dominante ligado ao cromossomo X, de penetrância e expressão variáveis, levando à heterogenicidade genética. Este defeito é que levaria a alterações bioquímicas na composição da membrana basal glomerular, cápsula do cristalino e algumas partes do ouvido interno.
RELATO DO CASO
Apresentamos o caso de S.B.M., 18 anos, sexo masculino, com história de hematúria macroscópica recorrente de início aos 5 anos de idade, sem queixas auditivas ou visuais. Foi submetido a exames de sangue e urina na época e diagnosticado glomerulonefrite, sem classificação quanto a seu tipo histológico. O quadro de hematúria macroscópica persistiu até os 14 anos, quando o paciente passou a ser assintomático.
Aos 10 anos o paciente começou a notar diminuição da acuidade visual e auditiva; com piora de ambos, a partirdos 15 anos, procurou os serviços de ORL. e OFT. da Faculdade de Medicina de Jundiaí, onde foi submetido a interrogatório específico, exames complementares nas respectivas áreas. Os resultados obtidos foram os seguintes:
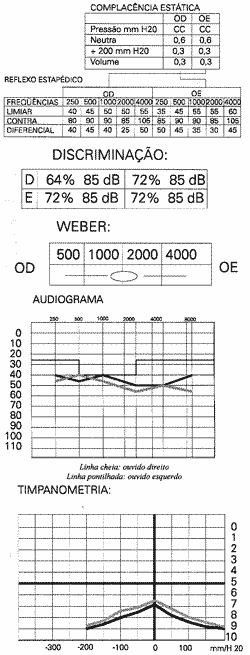
Avaliação ORL, paciente queixava-se de hipoacusia bilateral sem fator acompanhante ou desencadeante. Negava tinitus, vertigens, otaigia, otorréia, trauma craniano, uso de drogas ototóxicas ou antecedentes pessoais de patologia auditiva. A audiometria mostrava disacusia neuro-sensorial leve com curva plana bilateral, recrutamento em todas as freqüências e diminuição da discriminação vocal.
REFLEXO ESTAPÉDICO
Avaliação OFT, paciente queixa-se de baixa acuidade visual bilateral. Ao exame biomicroscópico apresentava lenticone anterior em olho direito, acuidade visual de 0,4/0,3, ceratometria de 43,75/44,75 50 em OD e 43,75/45,50 115 em OE. Foi feito diagnóstico de alteração do cristalino a esclarecer.
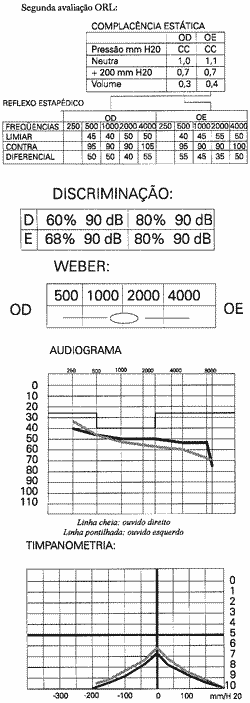
O paciente abandonou o acompanhamento clínico, mas com piora progressiva da sintomatologia nos últimos 3 anos, principalmente visual, voltou a procurar-nos para reavaliação. As queixas permaneceram as mesmas, tanto visual quanto auditiva, só que em grau mais avançado. Não havia queixa de alterações renais. Os resultados audiométricos apresentavam pouca piora em relação ao anterior, mas progressão do quadro oftalmológico.
Segunda avaliação OFT: Na biomicroscopia a córnea sem alterações, cristalino com aumento de curvatura anterior e posterior em forma de cone mais intenso à direita, opacidade subcapsular posterior ++14+ emOD, opacidade subcapsular anterior +++/4+ e posterior +/4+; ceratometria 43,75/44,75 110 em OD e 45,00/ 44,00 110 em OE; com refração de +2,00 DE A -1,75 DC 75 em OD e +1,75 DE A -1,00 DC 180 apresentava acuidade visual de 0,3/0,5.
Confirmamos assim a piora da acuidade visual pela acentuação da formação do lenticone que, além de ser bilatera,l é mais acentuada à direita, acomete tanto a cápsula anterior quanto a posterior do cristalino. Desenvolveu também catarata.
TIMPANOMETRIA:
Com base nesses resultados, foi feita a hipótese diagnóstica de Síndrome de Alport e encaminhamos o paciente para avaliação pelo serviço de nefrología. Sua única queixa era fraqueza. Ao exame físico não apresentou alterações significativas. Os resultados dos exames laboratoriais confirmaram a presença de hematúria microscópica, proteinúria, cilindras hialinos e granulosos; creatinina sérica aumentada e diminuição do clearance de creatinina urinária.
O quadro é compatível com alterações renais do tipo nefrítico, apesar do paciente assintomático.
DISCUSSÃO
Analisando os dados da história clínica, exames físico e laboratorial e avaliação oro-oftalmológica, achamos os resultados suficientes para enquadrar o paciente como portador da síndrome de Alport, já que apresenta a tríade clássica de disacusia neurosensorial progressiva, lenticone anterior e glomerulonefrite. Apesar de muitos autores indicarem a biópsia renal para confirmação diagnóstica, encaramos este procedimento invasivo e de alto custo financeiro, pois apenas à microscopia eletrônica podese detectar alterações precoces, sendo que nenhuma delas é patognomônica desta síndrome.
Por outro lado, a biomicroscopia, a audiometria tonal e a impedanciometria são exames de fácil realização ao nível de ambulatório e métodos não invasivos, cujos resultados são fundamentais para a conclusão diagnóstica de síndrome de Alport.
Não há tratamento específico. Já foi descrito o uso de imunossupressores sem melhora da evolução da doença.
O lenticone anterior é possível de correção cirúrgica através de facectomia e implantação de lente intraocular, mas os riscos de insucesso cirúrgico, devido a uma fragilidade maior da zônula, devem ser bem analisados na avaliação pré-operatória. Lentes corretivas são uma outra opção com resultados satisfatórios.
O uso de prótese auditiva é o tratamento de escolha nos casos de disacusia grave e especial atenção deve ser dada a pacientes com deficiência acentuada, a fim de mantê-los em atividade social próxima à normal.
Deve-se orientar o paciente quanto ao uso de drogas atotáxicas, principalmente utilizadas no tratamento renal, como aminoglicosídeos, furosemida, etc., que, aliadas à alterações metabólicas, agravariam a cocleopatia. O prognóstico da doença é reservado, já que a evolução é progressiva, levando a insuficiência renal terminal. O controle clínico é realizado através de dieta, dïuréticos e controle da uremia. A diálise é preconizada nos estágios avançados e o transplante renal, quando possível, pode levar à melhora da hipoacusia.
A evolução clínica do paciente em questão não teve grande evolução nos últimos três anos como esperado, apesar do mesmo ser do sexo masculino e apresentar proteinúria. Atualmente a hematúria é microscópica, compatível com os dados da literatura. Entre os familiares, apenas um irmão havia apresentado hematúria microscópica e queixas visuais compatíveis com miopia, sendo todos os outros familiares assintomáticos.
CONCLUSÃO
Através deste estudo, concluímos que o diagnóstico da síndrome de Alport pode ser basicamente clínico, desde que o paciente apresente os sinais e sintomas clássicos, reservando-se a biópsia renal para os casos duvidosos ou onde há a necessidade do estudo familiar em indivíduos assintomáticos.
A cocleopatia parece realmente ser devida ao acúmulo de metabólitos no organismo, já que houve melhora da disacusia em pacientes submetidos a transplante renal.
Ressaltamos mais uma vez a importância da orientação a esses pacientes quanto ao uso de drogas ototóxicas e avaliação auditiva periódica e, se necessário, o uso de prótese auditiva na tentativa de assegurar a qualidade de vida e convívio social.
REFERÊNCIA BIBLIOGRÁFICA
1. Alport, A. C.: "Hereditary familial congenital haemorragic nephritis". Brit. Med. J., 1: 504, 1927.
2. Catandi, M. D.: "Alport Syndrome". Ear, nose and throat journal, 68 nov. 1989.
3. Zavala et al.: "Nefropatia crônica hereditária con hipoacusia y defectos oculares"
5. Sohar, E.: "A heredofamilial syndrome caracterized by renal disease, inner ear deafness and ocular changes", Harefuah, 47: 161-6, 1954.
6. Perkoff, G. T. et al.: " Clinical study of hereditary intersticial pyelonephritis", Arch, int. med., 88: 191, 1951.
7. Nielsen, C.E.: "Lenticone anterior and Alport syndrome", Acta ophtalmo., 56: 518-30,1978.
8. Brownell, R. D. et al.: "Anterior lenticones in familial hemorrhagic nephritís: demonstration of tens pathology", Arch opht., 71: 481-3,1964.
4. Reznik, M. V. et ai.: "Hereditary nephritis and sensorineural deafness", Journal of pediatrics, pg 676, 1988.
9. Hinglais, N. et al.: " Characteristic ultraestructural lesion of glomerular basement membrane in progressive hereditary nephritis", Lab. invest., 27:473-87, 1972.
10. Yoshikawa, N. et al.: " The glomerular basal lamina in hereditary nephritis", J. pathol., 135: 199-209, 1981.
11. Yun, M. et al.: "Basement membrane nephropaty: a new classification of Alport's syndrome and assyntomatic hematuria based on ultraestrutural findings". Hum. pathol., 14: 996-1003, 1983.
Dept° ORL 1 OFr Fac. Medicina Jundiaí Av. dos Coqueiros S/N
Franco da Rocha - São Paulo
Apresentado: IX Reunião da Sociedade Brasileira de Otogolia 1 Jornada Amazônica de Fonoaudiologia
Dr. Paulo H.A. Morales
R. Engenheiro Sá Rocha n° 345 Pinheiros São Paulo - SP CEP 05454-020
Artigo aceito em 15 de outubro de 1993.
Imprimir: ![]()