

Ano: 2000 Vol. 66 Ed. 3 - Maio - Junho - (11º)
Seção: Relato de Casos
Páginas: 273 a 276
HOLOPROSENCEFALIA COM PROBÓSCIDE - CASO CLÍNICO.
Holoprosencephly with Proboscis A Clinical Report.
Autor(es):
Ney P. Castro Jr.*,
Lídio Granato*,
Marina S. Figueiredo**,
Oswaldo A. B. Rios***.
Palavras-chave: holoprosencefalia, probóscide, audiometria de tronco cerebral
Keywords: holoprosencephaly, proboscis, brain stem electric response
Resumo:
A holoprosencefalia é o conjunto de displasias crânio-faciais e encefálicas, originárias da clivagem anômala do prosencéfalo. A displasia do andar médio da face, principalmente dos olhos e da pirâmide nasal, são variáveis e refletem a severidade da displasia encefálica. A freqüência estimada da holoprosencefalia é de 1:15:000 nascimentos. A raridade desta displasia, as disgenesias da pirâmide e -cavidade nasais e a associação com malformações encefálicas justificam a apresentação de um caso clínico desta entidade. Trata-se de um rescém-nato, do sexo feminino, proveniente de segunda gestação a termo e com peso corporal adequado à idade gestacional. Ao exame físico inicial, apresentava várias anomalias craniofaciais: hipoteleorbitismo com microftalmia e estrutura nasal rudimentar, uma probóscide. O exame por imagem do crânio, pela tomografia computorizada, foi confirmatório para holoprosencefalia alobar e probóscide associado à atresia coanal. O cariótiopo foi 46, XX. A avaliação audiológica objetiva, feita aos três meses de idade, revelou emissões otoacústicas essencialmente normais. A audiometria de tronco encefálico revelou presença de P-I e P-III, com LIP P:I-III aumentadas. Estes dados confirmam o comprometimento auditivo funcional de tronco encefálico, compatível com o diagnóstico por imagem de holoprosencefalia alobar.
Abstract:
Holoprosencephaly is a group of craniofacial derangement, from impaired midline cleavage of the embrionic forebrain. The midline facial developmental anomalies, mainly of the eyes and nose are variable and usually reflect the severiry of underlying brain malformation. The frequency of holoprosencephaly is about 1:15000 live births. A clinical report of holoprosencephaly is presented, with comments about the malformations of the nose, central nervous system and an audiologic evaluation by evoked otoacoustic emissons and brain stem electric response. The proband was a girl born after an uneventful pregnancy. The physical examination at birth showed moderate respiratory distress and congenital facial anomalies: hypotelorbitism associated with mild microphtalmia; a rudimentary nasal structure (proboscis) located under the midline orbit, with one nostril and a virtual nasal cavity, without expiratory air flow. Further examination showed no other abnormalities. A CT scan confirmed the holoprosencephaly associated with choanal. The cariotype of peripherical blood was normal: 46,XX. She was submitted to evoked otoacoustic emisson (EOA-DP) and brain stem electric response (BSER) in order to evaluate auditory dysfunction. The EOA-DP presented normal responses and the BSER showed waves P-I through P-III with increased interpeak latencies, with absenc e of P-IV and P-V waves. These responses suggest a severe auditory dysfunction at the 1 high level of brain stem. The patient described fulfills the clinical diagnosis of cebocephaly, accorging to DeMeyer's classification of holoprosencephalie disorders. Due to poor central nervous system function and prognosis, we have choosen a conservative treatment.
![]()
INTRODUÇÃO
A holoprosencefalia é o conjunto de displasias cranio-faciais e encefálicas, originárias da clivagem anômala do prosencéfalo. Inicia no embrião entre as terceira e sexta semanas de gestação, produzindo fetos com malformações oculares e nasais associadas à malformações encefálicas2. As alterações do SNC potencialmente produzem disfunções do sistema auditivo, que podem ser detectadas pela audiometria de tronco encefálico.
As malformações oculares variam da ciclopia, caracterizada por estruturas oculares rudimentares, com ausência de nervos ópticos, no centro da fronte, à microftalmia associada ao hipoteleorbitismo2.
O andar médio da face é marcado pela displasia da pirâmide nasal, do complexo etmoidal e da pré-maxila. A pirâmide nasal, rudimentar, abriga uma cavidade nasal, única, desprovida do septo nasal e quase virtual; a atresia coanal pode estar presente. Na cavidade oral pode ocorrer a palatosquise e é comum a ausência dos incisivos centrais superiores. A displasia facial é proporcional à displasia encefálica2, 1, 4.
Encéfalo alobar manifesto por hemisférios cerebrais totalmente fusionados, sistema ventricular único e amplo, fusão de tálamo e ausências de corpo caloso e de seio sagital, ocorrem nas displasia faciais mais graves. A holoprosencefalia lobar e ou semilobar apresenta com clivagem parcial dos hemisférios cerebrais e sistema ventricular dilatado e com morfologia próxima da normal; ocorrem associados à manifestações faciais menos severas2, 4.
A freqüência estimada da holoprosencefalia é de 1:15.000 nascimentos.
Esta displasia pode estar associada à alterações do cariótipo como a trissomia 13 e a triploidia; aberrações cromossômicas envolvendo os cromossomos 2,3,7,18 e 21. Na literatura são relatados casos familiares, com forma de transmissão autossômica2, 1, 4.
É descrito neste trabalho um caso clínico de holoprosencefalia alobar, no qual foi avaliada a função auditiva através da audiometria de tronco encefálico. A raridade desta displasia, as disgenesias da pirâmide nasal do complexo etmoidal e oculares e a associação com malformações encefálicas justificam a apresentação de um caso clínico desta entidade. O resultado da avaliação auditiva pela audiometria de tronco encefálico provavelmente é a única descrita na literatura, neste tipo de afecção, segundo conhecimento dos autores.
APRESENTAÇÃO DE CASO CLÍNICO
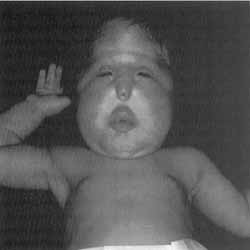
Trata-se de um rescém-nato, do sexo feminino, proveniente de segunda gestação a termo e com peso corporal adequado à idade gestacional. Ao exame físico inicial, apresentava hipotonia generalizada, moderado desconforto respiratório e várias anomalias craniofaciais: hipoteleorbitismo com microftalmia e estrutura nasa rudimentar caracterizada pela probóscide.
Figura 1. Neonato de quatro semanas de idade cronológica portador de próbóscide e hipoteleorbitismo.
A probóscide consistia de narina única, sem repto nasal, cavidade nasal virtual e sem fluxo nasal expiratório. Sem outras malformações corporais (Figura 1).
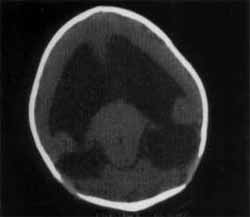
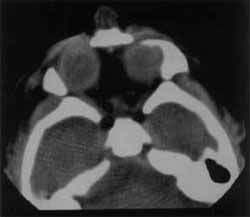
O estudo por imagem do crânio, pela tomografia computorizada e ultrassonografia transfontanela revelaram parênquima encefálico periférico e predominante na região frontal; um único sistema ventricular, arredondado e ocupando a área do parênquima cerebral; tálamos fundidos e ausência de corpo caloso; tronco encefálico aparentemente íntegro. A cavidade nasal, praticamente virtual, apresentava atresia coanal e teto esfenoidal baixo, estreitando o rinofaringe. O exame foi confirmatório para holoprosencefalia alobar e probóscide associado à atresia coanal (Figuras 2 e 3).
O cariótiopo do sangue periférico revelou-se normal: 46, XX.
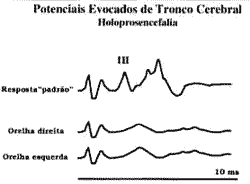
Foi executada avaliação auditiva, que revelou emissões otoacústicas - produto de distorsão essencialmente normais, entre 250 Hz a 4 kHz, em ambos os ouvidos. A audiometria de tronco encefálico revelou presença de P-I e P-III, com ausência de P-V; as LIP P:I-III encontram-se aumentadas de 4,2 ms (orelha direita) e de 4,1 ms (orelha esquerda) (Figura 4).
Apresentou sobrevida até os três meses de idade, com respiração bucal e deglutição expontânea; do ponto de vista neurológico apresentou hipotonia, crises convulsivas e evoluiu com hidrocefalia e macrocefalia. Teve óbito domiciliar por provável insuficiência respiratória aguda.
Figuras 2. Tomografia computadorizada de encéfalo, demonstrando cebocefalia alobar: o telencéfalo, representado por cortex cerebral delgada, principalmente na área frontal, e o restante preenchido por ventrículo de grandes dimensões.
Figura 3. Tomografia computadorizada de órbita e cavidades nasais, demonstrando hipoteleorbitismo e cavidades nasais com espaço virtual.
DISCUSSÃO
A holoprosencefalia ainda que rara, apresenta alta freqüência em embriões humanos abortados expontaneamente, 1:250 casos, cerca de 60 vezes a mais que os nascituros4. Apresenta vários graus de severidade de displasia, que afetam o encéfalo e a face; no caso apresentado, é denominado cebocefalia da classificação de DeMYER e colaboradores (1964) modificado por Elias, D. e colaboradores (1991)2.
Podem estar associadas a alterações cromossômicas, principalmente a trissomia do 13 e eventualmente envolvendo os cromossomos 2,3,7,18 e 212, 1, 4. A associação da holoprosencefalia com fatores externos tais como rubéola e toxoplasmose gestacionais, diabetes melito materna, drogas com efeito teratogênico são citadas na literatura, mas sem comprovação segura2, 4. Os pais do paciente em questão são saudáveis e a gestação ocorreu sem intercorrências.
Recentemente, a holoprosencefalia foi associada à sindrome de Charge: coloboma, cardiopatia, atresia coanal, retardo de crescimento e mental, hipogonadismo e malformação auricular com deficiência auditiva. O estudo por imagem do crânio e encéfalo, através da tomografia computadorizada e da ressonância magnética na síndrome de Charge apresentou uma prevalência5, 6.
O comprometimento neurológico do paciente não permitiu uma avaliação audiológica de observação comportamental. A emissão oto-acústica - produto de distorção - revelou ser essencialmente normal em ambos os ouvidos, atestando a integridade da cóclea.
Figura 4: Audiometria de tronco encefálico evidencia a presença de P-I e P-III, com LIP: I-III aumentada e ausência de P-V, sugerindo lesão do sistema auditivo em tronco encefálico alto.
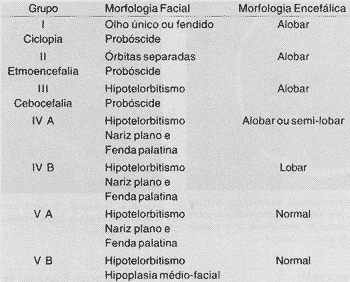
A audiometria de tronco encefálico revela em ambos as orelhas a presença de P-I e P-III, com suas latências interpicos aumentadas, e a ausência de P-V. Estes ciados confirmam integridade do sistema auditivo, das sinapses neuroniais da cóclea até o complexo olivar superior, no tronco encefálico baixo e comprometimento de tronco encefálico alto, compatível com o diagnóstico por imagem de holoprosencefalia alobar. Um estudo comparando os resultados da audiometria de tronco encefálico com ressonância magnética de encéfalo em crianças portadoras de paralisia cerebral espástica demonstrou a presença de potenciais auditivos mesmo nos casos com extensas lesões do telencéfalo e mesencéfalo3.TABELA 1 - Classificação de anormalidades holoprosencefálicas apud DeMyer e colaboradores (1964), modificado por Elias, D. e colaboradores (1991).
O estadiamento da holoprosencefalia pela classificação de De-Myer e colaboradores (1964), modificado por Elias, D. e colaboradores (1991), permite estabelecer o prognóstico e o planejamento da terapia a ser proposta. É indispensável o aconselhamento genético, devido à ocorrência de casos familiares2 (Tabela 1).
Os pacientes portadores de ciclopia, etmocefalia e cebocefalia (grupos de I a III) não sobrevivem além da infância; e, por este motivo, a cirurgia é contraindicada. Os pacientes do grupo IV A, podem sobreviver por anos, apesar de severos déficits neurológicos, e raramente são beneficiados pela cirurgia.
Os pacientes dos grupos IV B a VB devem ser considerados eletivos para a correção cirúrgica. Os pacientes do grupo IV B são mentalmente deficientes, mas com longa expectativa de vida, o que justifica a correção cirúrgica do lábio e palato fendidos.
Desta forma, a proposta da correção cirúrgica é apoiada no grau de diferenciação encefálica, na expectativa do período de vida e nas possibilidades de reconstrução facial. A investigação do cariótipo e aconselhamento genético são partes obrigatórias da investigação de tais casos, visto que alterações cromossômicas e mecanismos de transmissão genética são freqüentes nestes casos.
REFERÊNCIAS BIBLIOGRÁFICAS
1. COLLINS, A. L.; LUNT, P. W.; GARRETT, C.; DENNIS, N. R.: Holoprosencephaly: a family showing dominant inheritance and variable expression. J. Med. Genet., 30: 36-40; 1993.
2. ELIAS, D. L.; KAWAMOTO, H. K.; WILSON, L. F. - Holoprosencephaly and midline facial anomalies: redefining classification and management. J. Plast. Reconstr. Surg., 35: 951-8; 1992.
3. FOBE, L. P. O. - Estudo da correlação entre potencial evocado auditivo de tronco cerebral e ressonância magnética de crânio em crianças com paralisia cerebral espástica. São Paulo, 1988. 107 p. Tese (Doutorado) - Facudade de Medicina, Universidade de São Paulo.
4. GALGERA, M.; MALAVE, L.; LEON, A. - Hoploprosencephaly with proboscis. J. Ginec. Obstr. Investig., 42: 70-2; 1996.
5. HARRISJ; ROBERT, E.; KÄLLÉN, B. - Epidemiology of choanal atresia with special reference to the CHARLE association. Pediatrics, 99: 363-367; 1997.
6. REJJAL, A.; ALAIYAN, S.; COATES, R.; ABUZEID, M. - The prevalence and spectrum of brain abnormalities in congenital choanal atresia. J. Neuropedicatrics, 25: 85-8; 1994.
* Professor Doutor Adjunto e Chefe de Clínica do Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
** Professor Assistente do Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
*** Residente do 3° Ano do Departamento de Otorrinolaringologia da Santa Casa de São Paulo.
Trabalho realizado no Departamento de Otorrinolaringologia da Santa Casa de Misericórdia de São Paulo.
Apresentado na Secção de Temas Livres do XXXIV Congresso Brasileiro de Otorrinolaringologia, em Porto Alegre/ RS.
Endereço para correspondência: Rua Itapeva, 500, cjto.10-B - 01332-000 São Paulo/ SP - Telefone/Fax: (0xx11) 289-6048 - E-mail: neypcjr@ibm.net
Artigo recebido em 25 de junho de 1999. Artigo aceito em 20 de agosto de 1999.
Imprimir: ![]()