

Ano: 1982 Vol. 48 Ed. 3 - Julho - Setembro - (3º)
Seção: Artigos Originais
Páginas: 32 a 38
MALFORMAÇÕES CONGÉNITAS DA FACE UMA REVISÃO DAS SÍNDROMES MAIS IMPORTANTES
Autor(es): CARLOS A. OLIVEIRA, PhD *
Resumo:
Os fatos principais referentes a seis síndromes principais de malformações congênitas da face foram revistos. A ênfase foi colocada na etiopatogenia das manifestações clinicas. As conquistas passadas e os objetivos futuros no campo da pesquisa do desenvolvimento humana foram discutidos.
![]()
SUMÁRIO
Sera revista a literatura referente à Disostose Mandtbulo-Facial (síndrome de Treacher-CollinsFranchescetti), Microsomia Hemifacial (síndrome do primeiro e segundo arcos branquiais), Disostose Crânio-Facial (doença de Crouzon), Displasia Crânio-carpo-tarsal (síndrome da face de Assobio), Acrocefalossindactilia (síndrome de Apert) e Displasia Óculo-aurículo-vertebral (síndrome de Goldenhar). Os principais fatos relativos a cada uma destas síndromes serão apresentados com ênfase nos mecanismos embriológicos atualmente aceitos como causadores destas malformações faciais. Estas síndromes naturalmente não cobrem todas as malformações faciais congênitas desde que muitas combinações atípicas de malformações faciais ocorrem esporadicamente. O conceito de síndromes congênitas, suas limitações e a necessidade de conhecimentos mais sólidos sobre a etiopatogenia destas malformações serão discutidos nesta comunicação.
A observação e registro de fenõmenos naturais constitui o fundamento das ciências da natureza. O próximo passo para que seja gerado o conhecimento científico é o da organização dos fenõmenos observados em uma estrutura
lógica para a partir daí se chegar à criação de um método de investigação que caracterizará o rumo da Ciência.
Uma síndrome é um grupo de sinais e sintomas que freqüentemente ocorrem em conjunto. É basicamente um conceito clínico e não implica em nenhum conhecimento de etiopatogenia. E portanto uma primeira tentativa de organizar o conhecimento factual. Se for possível encontrar uma causa comum para estes sinais e sintomas, uma doença será definida e o tratamento dirigido para a correção dos mecanismos causais.
Quando tratamos de malformações congênitas da face, falamos de síndromes porque os mecanismos etiopatogênicos são desconhecidos ou hipotéticos. Entretanto foi possível definir algumas grandes síndromes que resistiram ao teste do tempo e portanto devem ter causas específicas. Na verdade, pesquisa dirigida no sentido de achar tais causas está começando a dar resultados.
Nos parágrafos seguintes, vamos apresentar os fatos conhecidos sobre as síndromes mais importantes de malformações faciais e comentar as deficiências no nosso conhecimento atual bem como as tendências futuras no campo da pesquisa nesta área.
I - DISOSTOSE MANDÍBULO-FACIAL (SÍNDROME DE TREACHER - COLLINS - FRANCHESCETTI).
Berry em 1889 descreveu dois casos, mãe e filha, com fendas palpebrais oblíquas e descendentes de dentro para fora; micrognatia, prolongamento da linha de implantação do cabelo na direção da região bucinadora e colobomas nas pálpebras inferiores. Em 1900 Treacher Collins descreveu um menino de oito anos e um homem de 32 anos com estigmas semelhantes. Foi Franchescetti em 1949 que pela primeira vez juntou todas as características que formam a síndrome e definiu-a como tal.
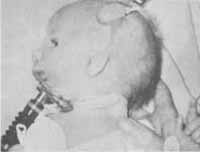
CARACTERISTICAS CLINICAS (Fig. 1)
a) Forma completa - obliquidade antimongolóide das fendas palpebrais, colobomas no terço lateral das pálpebras inferiores, falta de cílios nos dois terços mediais das pálpebras inferiores. Os malares são severamente hipoplásticos e todos os ossos derivados dos processos maxilar e mandibular do primeiro arco branquial mostram deficiências no potencial de crescimento. A maxila é defeituosa e o palato pode ser alto ou estreito ou apresentar uma fenda. A mandíbula é afetada principalmente ao nível do processo condilóide. A hipoplasia severa deste processo resulta em micrognatia. Os defeitos de maxila e da mandíbula resultam em má-oclusão. Além disto há malformações do ouvido externo e do ouvido médio, fístulas cegas entre o ângulo da boca e o ouvido e avanço da implantação de cabelo na direção da região geniana.
b) Forma incompleta - Esta forma tem todas as características citadas acima em um grau atenuado de tal sorte que a face parece mais normal.
Figura 1 - Síndrome de Treacher-Collins-Franchescetti. Veja descrição no texto.
c) Forma abortiva - Somente as deformidades das pálpebras estão presentes.
Franchescetti (2) descreveu uma forma unilateral da síndrome e uma forma atípica. A forma unilateral provavelmente representa casos de microsomia hemifacial e a forma atípica não pode ser aceita como pertencentes à síndrome em questão.
ANOMALIAS ASSOCIADAS
Anomalias das extremidades são descritas mais freqüentemente em associação com as anomalias faciais descritas. Aplasia bilateral de polegar e sinostose rádio-unlar (2), pé varo (3), segundo metatarso anormalmente longo (4), sindactilia bilateral parcial dos artelhos (5), união membranosa entre os dedos da mão e dos pés (6), klinodactilia (7), articulações metacarpofalangeanas e metatarso-falangeanas hiperextensíveis (4) ocorrem em alguns casos da síndrome.
Assimetria facial (8), hidrocéfalo interno (2), anquilose da articulação temporomandibular (5), ausência da apófise mastóide (2), ausência da parótida (9), deficiência mental (3) e linfagioma do lábio superior (10) foram descritos em associação com a síndrome em questão. Descritos menos freqüentemente são atresia coanal (11), tórax assimétrico(6), deformidades do esterno(12), fusão das vértebras (2), hidronefrose (13) e anomalia na descida dos testículos (5).
As anomalias otológicas merecem atenção especial ao descrevermos esta síndrome. Cerca de 48°%
dos casos descritos na literatura é de surdez (1). As anomalias são em geral restritas aos ouvidos externo e médio e variam de completa ausência do pavilhão auricular-e conduto auditivo externo a pequenos defeitos no pavilhão auricular. Anomalias dos ossículos no ouvido médio são freqüentes. Anomalias dos canais semicirculares laterais foram descritas em conexão com esta síndrome (14).
ETIOPATOGENIA
A síndrome é transmitida geneticamente como traço autossômico dominante e a incidência é igual nos dois sexos (1:1) (15). A variabilidade nas deformidades de caso para caso deve ser explicada assumindo que se trata de um gene instável de expressão, penetrância e especificidade variáveis( Poswillo (16) descreveu um modelo animal para a síndrome de Treacher-Collins-Franchescetti. Ele reproduziu a síndrome em uma cepa de ratos administrando 100.000 IU de vitamina A solúvel em água no dia, 8/5 de desenvolvimento fetal à rata grávida. Cem por cento dos fetos desenvolveram a síndrome. Ele observou ainda que o mecanismo etiopatogênico envolvido na produção das deformidades é destruição precoce das células da crista neural dos primórdios da face e do ouvido (externo e médio) que migrariam para o primeiro e para o segundo arcos branquiais. Baseado nestes experimentos ele propõe uma teoria para explicar a produção das deformidades.
II - MICROSSOMIA HEMIFACIAL
Esta síndrome ocorre uma vez em cada 3.500 nascimentos bem sucedidos (feto vivo), não tem preferência por nenhum sexo (1:1) e não foi identificado até aqui nenhum fator genético.
CARACTERÍSTICAS CLÍNICAS
As anormalidades são usualmente unilaterais e consistem de redução do pavilhão auricular e do trago, irregularidades da cadeia ossicular, hipoplasia só côndilo mandibular, do ramo ascendente da mandíbula e do malar, deficiência do masseter, do músculo temporal e dos músculos pterigóides e paresias dos músculos de expressão facial. Conseqüentemente a característica marcante é assimetria facial. Se as deformidades fossem bilaterais seria muito difícil separar esta síndrome da descrita anteriormente.
ETIOPATOGENIA
Poswillo em 1972 produziu as deformidades que estão presentes na síndrome humana em roedores injetando a droga Triazene (1,Smgm diluída em solvente oleoso) intrauterinamente no décimo primeiro dia de gestação. A incidência das deformidades foi de 100% em ratos da cepa Wistar. O exame histopatológico dos fetos mostrou evidência de hematomas ao nível da junção das artérias faringeana ventral e hioidiana. Ele postulou que estes hematomas ocorrendo durante o desenvolvimento embriológico na área do primeiro e do segundo arcos branquiais são responsáveis pela produção das deformidades presentes no nascimento. O hematoma surge em conseqüência de vazamento nas anastomoses dos vasos que formam o tronco da artéria estapediana. A variabilidade no tamanho dos hematomas seria responsável pela variabilidade das deformidades finais. Do mesmo modo a expansão do hematoma além dos limites do primeiro e segundo arcos explicariam o envolvimento de estruturas não derivadas deles. A causa exata do vazamento não ficou clara nestes experimentos. Para explicar a ocorrência de deformidades associadas em outras áreas do corpo ele postulou a ação de algum agente teratogênico em diferentes partes do embrião. Que agente teratogênico causaria a síndrome permanece uma incógnita (17).
III - DISOSTOSE CRANIOFACIAL (DOENÇA DE CROUZON)
Esta síndrome é causada pelo fechamento prematuro de algumas suturas do crânio e da face. Crouzon em 1912 descreveu pela primeira vez as características clínicas e descreveu 15 casos em 20 anos. (18)
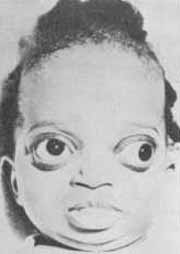
CARACTERÍSTICAS CLÍNICAS (Fig. 2)
A doença de Crouzon faz parte de um grupo de malformações congênitas caracterizadas por fechamento prematuro de suturas cranianas. Ela é diferenciada da cranioestenose simples pela presença de malformações faciais associadas. A síndrome consiste de: 1) Deformidades cranianas (a deformidade específica depende de que suturas são fechadas prematuramente); 2) Exoftalmos e hipertelorismo (Fig. 2); 3) Hipoplasia maxilar e conseqüente prognatismo relativo; 4) Palato arqueado (alto) e 5) Perfil de "bico de papagaio".
ANOMALIAS ASSOCIADAS
Nistagmo, estrabismo e cegueira podem ocorrer em conseqüência de alta pressão endocraniana. Atresia das coarias e atresia bilateral dos condutos auditivos externos já foram descritas. Anomalias do ouvido médio são vistas ocasionalmente.
Em alguns casos a língua é aumentada de volume e protrusa entre os lábios. Anodontia parcial, dentes que não têm erupção normal e má-oclusão são características freqüentemente encontradas. Desvio do septo associado com respiração estertorosa são freqüentes e surdez aparece com relativa freqüência (19,20).
Figura 2 - Doença de Crouzon. Veja descrição no texto.
ETIOPATOGÉNESE
Há controvérsia com relação ao papel da hereditariedade na produção desta síndrome. Baldwin (20) afirma que ao contrário das outras sinostoses prematuras a doença de Crouzon tem um caráter hereditário definido em pelo menos 60% dos casos. Kushner et al (19) discutem a possibilidade de uma herança heterozigótica dominante mas admitem que ocorrem casos esporádicos sem antecedentes familiares. Existem algumas teorias etiopatogênicas; Park e Power acreditam que existe um defeito no mesênquima das suturas que se fecham prematuramente, e est teoria é a mais aceita atualmente.
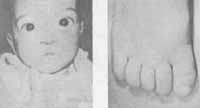
IV - ACROCEFALOSSINDACTILIA (SÍNDROME DE APERT) (Fig. 3)
Esta síndrome apresenta deformidades no crânio, face, mãos e pés. As deformidades crânio-faciais são semelhantes às encontradas na doença de Crouzon, e as síndromes são separadas pela ocorrência na acrocefalossindactilia de fusão dos dedos (sindactilia). É curiosa também a observação de que jamais as duas síndromes foram encontradas em membros da mesma família (21).
ANOMALIAS ASSOCIADAS
Rins policísticos, atresia do esõfago, estenose pilórica, ausência das fissuras pulmonares inferiores, atrofia de artérias pulmonares, aplasia pulmonar, útero bicornuado, ânus ectópico, duplicação de veia cava superior, estenose
pulmonar, defeito do septo interventricular, hidronefrose e hidronefrose e hidroureter, anomalias cartilaginosas da traquéia e brônquios, fibroelastose endocárdica, queratoconus e surdez podem acompanhar as anomalias principais (21).
Figura 3 - Síndrome d e A.Pert. Veja descrição no texto.
ETIOPATOGÉNESE
A síndrome ocorre em 1 de cada 100.000 a 160.000 nascimentos de fetos vivos (21) e acredita-se ser herdada como um traço autossômíco dominante com uma alta taxa de mutação. Também já foi relacionada a ocorrência da síndrome em decorrência da idade avançada dos pais. Não existem estudos experimentais sobre a etiopatogenia desta síndrome.
V-DISPLASIA OCULOAURICULOVERTEBRAL (SÍNDROME DE GOLDENHAR)
Características Clínicas (Fig. 4)
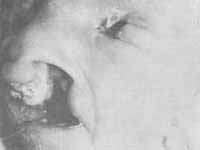
O crânio pode ser assimétrico-com bossa frontal. O cabelo pode ser implantado muito baixo, ao nível das sobrancelhas. A face parece de papagaio devido à hipognatia e hipoplasia malar. Atresia ou hipoplasia das narinas podem estar presentes. Pode haver uma microssomia hemifacial associada e neste caso a aparência da face é chocante. Nos olhos dermóides epibulbares ou lipodermóides são constantes. Os dermóides são a característica que diferencia esta síndrome da de Treacher-Collins e da microssomia hemifacial. Coloboma unilateral da pálpebra superoor é comum. Anoftalmia, microftalmia, microcórnea, coloboma da coróide ou da íris, atrofia da íris e catarata polar podem ocorrer. Microtia é comum. Surdez e(ou) ausência do conduto auditivo externo já foram descritos. Apêndices auriculares bilaterais são vistos constantemente e podem ser únicos, sésseis ou pediculados, mas são usualmente múltiplos. Eles em geral se localizam imediatamente em frente do trago ou ao longo de uma linha começando no trago e terminando no ângulo da boca (linha de fusão dos dois processos derivados do primeiro arco). Fístulas cegas são freqüentemente encontradas na mesma região. Manifestações músculo-esqueléticas são mais comuns na coluna vertebral. Occipitalização do atlas, vértebras cuneiformes, sinostose completa ou parcial das vértebras cervicais ou blocos de duas ou mais vértebras cervicais, vértebras supernumerárias torácicas ou lombares, hemivértebras, spina bífida, lombarização das primeiras vértebras sacras, aplasia de vértebras sacras, escoliose cérvico-torácica costelas anormais, fóvea sacral, sutura metópica acentuada, abertura no osso ao nível do vértex e fontanelas abertas são achados comuns. Manifestações neurológicas - Retardo mental, epilepsia e déficits motores foram descritos em uns poucos pacientes com esta síndrome. Como manifestações miscelânicas, anomalias da artéria renal e atresia anal foram descritas em portadores de síndrome.
Manifestações orais - Micrognatia é comum. Microssomia hemifacial pode ocorrer associada com esta síndrome. Hipoplasia maxilar e palato arqueado foram descritos. Macrostomia, língua bífida, úvula bífida, freio lingual duplo, fenda labial e palatina (Fig. 4), filtro alargado, má-oclusão e malformações dentárias podem ocorrer (22).
Figura 4 - Síndrome de Goldenhar. Veja descrição no texto.
ETIOPATOGÉNESE
De acordo com Gorlin et al. (22) não existe evidência de um padrão hereditário para esta síndrome. Eles consideram a síndrome de Goldenhar como uma variante de microssomia hemifacial e consideram que as anomalias surgem em conseqüência de anomalias no suprimento sangüíneo para a cabeça e pescoço tal como descrito para a microssomia hemifacial. Eles não apresentam nenhuma explicação para as anomalias que ocorrem em outras áreas da economia.
Eles também não têm explicação para o coloboma de pálpebra superior nem para os dermóides epibulbares. "Outras avenidas deverão ser exploradas para explicar estas anomalias" (22).
VI - DISPLASIA CRÃNIO-CARPO-TARSAL (SINDROME DA "FACE DE ASSOBIO")
Freeman e Sheldon em 1933 (23) descreveram o primeiro caso desta síndrome. Desde então 24 casos apareceram na literatura com uma variedade de nomes diferentes (24).
CARACTERISTICAS CLÍNICAS (Fig. 5)
Face - Expressão facial fixa, como máscara. Olhos fundos, lábios pregueados, rugas profundas no queixo, filtro longo e bochechas cheias, microssomia, ponte nasal larga com colobomas das asas do nariz.
Figura 5 - Síndrome da "face de assobio". Veja descrição no texto.
Anomalias Associadas - Desvio uInar dos dedos, deformidades fixas de flexão dos dedos ao nível da articulação metacarpofalangeana e da interfalangeana proximal, polegares fixos em adução, deformidades eqüinovaros dos pés bilateral, coluna torácica chata e sem movimentos evidentes da caixa torácica quando respirando.
As anomalias faciais e digitais e uma estatura baixa no adulto são as únicas características constantes da síndrome. Metade dos casos descritos mostraram anomalias ósseas dos pés (24).
ETIOPATOGÉNESE
O modo de transmissão como um traço autossômico dominante foi estabelecido claramente em um bom número de famílias estudadas (24). Entretanto nove dos casos descritos até hoje não têm antecedentes nas famílias e são interpretados como mutações recentes. A variabilidade das manifestações são atribuídas à expressividade variável do gene responsável pela síndrome.
As anormalidades são compatíveis com a vida, e indivíduos com esta síndrome sobrevivem e chegam à vida adulta. Eles têm alta incidência de problemas respiratórios devido à baixa mobilidade da caixa torácica.
DISCUSSÃO
Estai síndromes são todas mais ou menos raras e, como Gorlin et al. (25) dizem, a primeira questão que nos passa pela mente é por que perder tempo com elas quando existem tantas outras doenças mais freqüentes e mais importantes exigindo nossa atenção.
Esta questão é plenamente respondida quando uma revisão da literatura pertinente nos mostra quanto o estudo destas "experiências da natureza" (25) tem contribuído para nosso conhecimento da Biologia do Desenvolvimento. Estes conhecimentos têm naturalmente implicações bem além dos limites da síndrome em si.
Depois de uma considerável confusão causada pela descrição de muitas síndromes diferentes que tiveram de ser descartadas posteriormente como combinações esporádicas de malformações que não aparecem freqüentemente juntas, parece-nos que agora temos algumas síndromes bem definidas que se produzem na natureza de modo consistente e que portanto devem admitir causas comuns. Em outras palavras, conseguimos um primeiro passo no sentido da sistematização de nossos conhecimentos, que nos permitem partir para o próximo passo na estruturação do conhecimento científico: a elucidação dos fatores causais envolvidos.
A análise genética de famílias portadoras de indivíduos afetados destas síndromes permitiu a identificação de padrões de transmissão hereditária para a maioria das síndromes discutidas acima. Estamos em condições de aconselhar efetivamente estas famílias no sentido de evitar o aparecimento de malformações nas gerações futuras. A Ciência pura e aparentemente alienada das realidades cotidianas permite-nos atuar efetivamente no "dia-a-dia" da medicina preventiva.
Devemos estar conscientes entretanto de que estamos muito longe de ter todas as malformações que ocorrem na face bem organizadas em padrões definidos. Muitas combinações "atípicas" ainda ocorrem e esperam sistematização.
É necessário continuar o trabalho em duas frentes: observar as "experiências da natureza" clinicamente e tentar organizá-las em padrões definidos e que se reproduzam freqüentemente e tentar reproduzir estes padrões no laboratório para aprendermos sobre suas causas (ouvir com atenção a natureza e em seguida interrogá-la inteligentemente).
De um ponto de vista prático seria altamente desejável que existissem centros para o estudo de malformações congênitas para onde convergiriam todos estes casos para serem observados por superespecialistas. Evidentemente muito mais seria aprendido deste modo. Um Simpósio recente do NIDR (National Institute for Developmental Research - USA) reconhece a utilidade potencial destes centros mas também reconhece as dificuldades práticas para sua criação imediata (26). O estudo das síndromes congênitas tem sido altamente gratificante até aqui e o futuro parece ainda mais promissor ú medida que mais peças do quebra-cabeças encontram seu lugar certo.
SUMMARY
The main facts about 6 major syndroms of congenital facial malformations have been reviwed. Emphasis has been placed in the etiopathogenesis of the clinical manifestations. Comments directed to past achievements in this field and to future goals for research have been presented.
REFERÊNCIAS
1. MARAN, A.G.D. - J. Laryngol Otol 78:135-151, 1964.
2. FRANCHESCETTI, A.; Klein, D. - Acta Ophtalm (Kbh), 27:143, 1949.
3. GRANRUD, H. - Acta Paed (Uppsala) 42: 499, 1953.
4. STRAITH, C. L. and LEWIS, J. R. - Plastic and Recomstr Surg 4:204, 1949.
5. BREGEAT, P. and NAUD, G. - Arch Ophtalm (Paris) 9:427, 1949.
6. RONCONI, G. - Lattante 33:71, 1962.
7. HERBERTS, G. - Acta Oto-Laryng (Stock) 54:457, 1962.
8. PIRES de LIMA, G. e MONTEIRO, H. - Arch AnatAntrop (Lisboa) 8:185, 1923.
9. MCKENZIE, J. -Arch. Dis. Chidh. 33:477, 1958.
10. HARRISON, M.S. - J. Laryng 71:597, 1957.
11. MCNEIL, K.A. e WYNTER-WEDDERBURN, L. - J. Laryng 67:365,1953.
12. MANN,1. e KILLNER, T.P. - British J. Ophtalm 27:13,1943.
13. LIVINGSTONE, G. - J. Laryng 72: 750, 1958.
14. SANDO, I.; HEMENWAY, W.G.; MORGAN, W. R. - Trans. Am. Acad. ~tal. and Otolaryng 72:913, 1968.
15. POSWILLO, D. - J. Maxillofacial Surg. 2:64, 1974.
16. POSWILLO, D. - British J Oral Surg, 1974b.
17. POSWILLO, D. - Oral Surg. Oral Med. Oral Pathol 35:302, 1973.
18. CROUZON, O. - Bull--et mem Soe Med I de Paris 33:545, 1912.
19. KUSHNER, G.; ALEXANDER, E. Jr.; DAVIS, C.H. Jr. et al - J'Neurosúrg 37:434, 1972.
20. BALDWIN, J.L. - Laryngoscope 78:1.660,1968.
21. BERGSTROM, L.; NEBLETT, L.M.; HEMENWAY, W.G. - Arch. Otolaryngol. 96:117,1972.
22. GORLIN, R.J.; JUE, K.L.; JACOBSEN, U.; GOLDSCHMIDT, E. - J. Pediatr. 63:991,1963.
23. FREEMAN, E.A.; SHELDON, J.H. -Arch. Dis. Chid. 13:277, 1938.
24. MCLEOD, P.; PATRIQUIN. H. - Clin. Pediat. 13:184,1974.
25. GORLIN, R.J.; CERVENKA, J.; PRUZNSKY, S. - Birth Defects 7(7):3, 1971.
26. CHRISTIANSEN, R.L.; EVANS, C.A. - Cleft Palate J. 12:167, 1975.
* Professor Colaborador Nível 1 V do Departamento de Medicina Especializada da Faculdade de Ciênciasda Saúde da Universidade de Brasília; Responsável pela Disciplina de Otorrinolaringologia.

Imprimir: ![]()
All rights reserved - 1933 /
2026
© - Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial