

Ano: 1995 Vol. 61 Ed. 5 - Setembro - Outubro - (8º)
Seção: Relato de Casos
Páginas: 400 a 412
ANGIOFIBROMA JUVENIL INTRACRANIANO COM INVASÃO MENÍNGEA E DE SEIO CAVERNOSO.
Intracranial Juvenile Angiofibroma With Dural and Cavernous Sinus Invasion.
Autor(es):
Ossamu Butugan*,
Luiz Ubirajara Sennes**,
Edigar Resende de Almeida***,
Aroldo Miniti****.
Palavras-chave: Angiofibroma juvenil, meninge
Keywords: Juvenile angiofibroma, dura
Resumo:
Angiofibromas são tumores vasculares benignos, de crescimento lento e silencioso. Quando diagnosticados tardiamente, geralmente apresentam extensa destruição das estruturas naso-sinusais, com envolvimento da nasofaringe, fossas pterigopalatina e infratemporal. Quando existe extensão intracraniana, ocorre deslocamento da dura-mater, sem invasão da mesma. Em tumores recidivados, as alterações ocasionadas pelo tratamento anterior podem proporcionar verdadeira invasão da meninge. Na experiência de 123 rasos de angiofibroma tratados no Serviço de Otorrinolaringologia do Hospital das Clínicas ria Faculdade de Medicina da Universidade de São Paulo, no período de 1967 a 1994, a invasão de dura-máter ocorreu somente em 3 casos, todos com comprometimento de seio cavernoso concomitante. Destes pacientes, um não havia sido submetido a qualquer tipo de tratamento prévio. Esses pacientes foram tratados com remoção cirúrgica parcial do tumor, seguida de radioterapia e hormonioterapia, com bons resultados. O objetivo deste trabalho é relatar nossa experiência com essa apresentação não usual, discutindo o diagnóstico da associação das modalidades terapêuticas para o tratamento dessas casos.
Abstract:
Angiofibroma is a rate vascular benign tumor; that grows slowly and silent. In advanced cases, an extensive destruction of the naso-sinusal estructures, and accometiment of nasopharynx, esphenopalatine and infratemporal fossa often can be seen. An intracranial extension can occur, displacing the dura, without invasion. On recurrent tumors, Me local alterations induced for the treatment could promove a real dura invasion. Between 1967 and 1994, 123 cases of juvenile angiofibroma were treated at the ENT Department of the University of São Paulo. Only 3 cases presented a dura invasion, all of then witlz cavernous sinus accometiment. Two of them were recurrent tumor, but one had no previous treatment. These patients were treated by parcial intracranial resection, following by radiation and hormone therapy, with success. The objective of this paper is to report our experience with this unusual presentation, and discuss the diagnosis and the treatment in this cases.
![]()
INTRODUÇÃO
Angiofibroma juvenil é tumor vascular benigno, que acomete geralmente adolescentes e adultos jovens do sexo masculino. É tumor raro, representando menos de 0,05% dos tumores da cabeça e pescoço. A ocorrência nos atendimentos otorrinolaringológicos varia de 1 em 5 0001 a 1 cm 50 000 casos2.
Histologicamente3, é composto por estroina de tecido conectivo, com matriz de estruturas vasculares dilatadas. Embora benigno, pode apresentar comportamento extremamente agressivo, podendo causar destruição óssea por compressão.
Sua irrigação ocorre basicamente através da artéria maxilar interna, que se dilata à medida que o tumor se desenvolve e requer maior aporte sanguíneo. Nesta fase, ocorre também o desenvolvimento de circulação colateral, tanto pelos tecidos adjacentes como por outros ramos arteriais, até mesmo ramos da carótida interna.
Quando diagnosticados tardiamente, geralmente apresentam extensa destruição das estruturas naso-sinusais, com envolvimento do nasofaringe, fossas pterígopalatina e infratemporal. A progressão deste crescimento pode levar à invasão infra-craniana, ocorrendo deslocamento da duramáter, sem invasão da mesma. A literatura faz pouca referência à possível invasão da dura-máter. Em tumores recidivados, a ocorrência de invasão dural é mais aceita, mas em pacientes sem nenhuma forma de tratamento prévio, essa invasão é. pouco aceita.
Na experiência do Serviço de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo, observamos a presença de invasão de dura-máter em três dos 123 casos tratados no período de 1967 a 1994. Um destes casos não Havia sido submetido a qualquer forma de tratamento prévio.
O objetivo deste trabalho é relatar nossa experiência com esta apresentação não usual, discutindo o diagnóstico e a associação das modalidades terapêuticas para o tratamento dos mesmos.
APRESENTAÇÃO DOS CASOS
CASO 1: F.A.O., 19 anos, masculino, branco, com história de obstrução e sangramento nasal de repetição há 5 meses; procurou nosso pronto-socorro com epistaxe severa, controlada com tamponamento anterior. Apresentava-se descorado (Hb = 7,4), sendo transfundido com 2 U de concentrado de hemáceas. Após retirada do tampão nasal, observou-se massa tumoral vinhosa em fossa nasal direita que abaulava o pálato isole, podendo ser visualizada no rinofaringe. Ambas membranas timpânicas estavam retraídas. Foi feita Hipótese. de angiofibroma juvenil, confirmado por TC, que mostrou massa hipercaptante extensa ocupando rinofaringe, com extensão para seio esfenoidal e fossas nasal, ptérigo-palatina e infratemporal direita, com acometimento da musculatura pterigóide e erosão das paredes posteriores do seio maxilar e das lâminas pterigóides direitas. A massa invadia a fossa média do crânio, através de falha óssea na região da fissura orbitária inferior direita. Angiografia mostrou irrigação pela artéria maxilar interna e faríngea ascendente direita, sendo realizada embolização com Gelfoam e comprovada a desvascularização do tumor. Após 9 dias, foi submetido a cirurgia por "degloving", sendo realizada remoção completa do tumor. Três dias após, foi removido o tampão, recebendo alta no 4°PO. O paciente permaneceu assintomático por 18 meses, quando voltou e foi visualizada nova massa tumoral em fossa nasal direita. A TC mostrou recidiva tumoral extensa, compromentendo cavidade nasal direita, rinofaringe, seios maxilar e esfenoidal, fossas ptérigo-palatina, infra-temporal e craniana média, e cavidade orbitária do mesmo lado. Tal massa destruía o septo nasal, as paredes do seio maxilar, asas pequena e grande do esfenóide, lâminas pterigóides e parte do osso temporal à direita. Arteriografia mostrou massa nutrida pela maxilar interna direita e ramos da carótida interna (segmento petroso/ cavernoso), meníngea média, faríngea ascendente, palatina ascendente e outros ramos da facial direita. Foram embolizadas com Ivalon 600-900 U as artérias meníngea média, maxilar interna e palatina ascendente direitas. Cinco dias após, foi submetido a craniotomia órbito-fronto-zigomática, encontrando-se o processo tumoral infra-craniano, que atravessava a dura-máter, estando em contato com o parenquima do lobo temporal direito. Foi ressecaria todo tumor intracraniano, exceto a porção tumoral em contato com a porção inferior do seio cavernoso. O paciente evoluiu com paralisia do VI par craniano e síndrome de Horner, compatível com lesão a nível de seio cavernoso. No 8°PO, desenvolveu meningite bacteriana, sendo tratado com Vancomicina e Rocefin por 18 dias, com controle do quadro, recebendo alta hospitalar. Vinte e um dias após, foi submetido a nova cirurgia através de acesso por "degloving", com retirada completa da porção extra-craniana do tumor. Após 5 dias, foi retirado o tamponamento nasal, recebendo alta Hospitalar no 7° PO. O tratamento foi complementado com radioterapia e Hormonioterapia. Atualmente, com 3 anos de acompanhamento, não apresenta sinais de recidiva da doença, e obteve regressão parcial das alterações oftalmológicas.
CASO 2, E.A.C., 10 anos, masculino, branco, procurou nosso Serviço com história de obstrução e sangramento nasal há 6 meses, apresentando massa em fossa nasal direita.
A TC mostrou extensa massa vascularizada da fossa ptérigopalatina direita, extendendo-se para fossa nasal, antro maxilar, cavidade orbitária, rinofaringe e seio esfenoidal. Ocupava e destruía a base do crânio, invadindo a fossa craniana média direita e região selar, em situação extradural. A angiografia mostrou que o tumor era nutrido pelas artérias maxilar interna e meníngea média, que foram embolizadas com fragmentos de Gelfoam, Havendo desvascularização do tumor. Seis dias após, foi submetido à ressecção da massa vira "degloving", recebendo alta no 7°PO. Cinco meses após, a TC de controle mostrou recidiva, ocupando rinofaringe, seio maxilar direito, células etmoidais c esfenoidais.
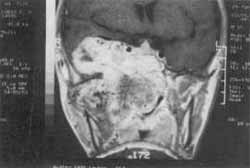
Figura I: Extensão do tumor inicial. Notar a extensão intracraniana invadindo e atravessando a dura-máter, estando em contato com o parênquima cerebral. Observar acometimento do seio cavernoso e artéria carótida interna (Caso 3).
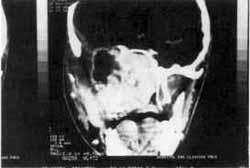
Figura II: Após-exérese parcial do tumor intracraniano. Notar o tumor residual junto ao seio cavernoso (Caso 3).
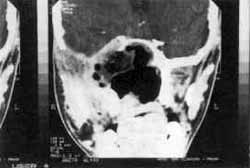
Figura III: Após exérese completa do tumor extracraniano. Notar tumor residual junto ao seio cavernoso (Caso 3).
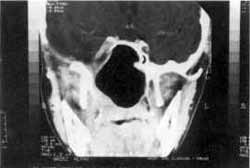
Figura IV: Após tratamento do tumor residual com hormônio e radioterapia (Caso 3).
Extendia-se para fossa temporal, fossa craniana média, sela túrcica e seio cavernoso. Angiografia mostrou nutrição por ramos da artéria maxilar interna direita e da carótida interna c artéria oftálmica. Foi realizada embolização seletiva dos ramos da carótida externa com partículas de Ivalon, havendo desvascularização parcial do tumor. Devido à grande participação dos ramos da carótida interna direita, foi colocado balão de oclusão abaixo da oftálmica e embolizado à jusante. O controle angiográfico mostrou devascularização parcial, restando apenas a nutrição proveniente da artéria oftálmica. O paciente evoluiu com amaurose do olho direito, pela presença de êmbolo ocluindo um ramo da artéria central da retina. Nove dias após a embolização, foi submetido à craniotomia fronto-órbito-têmporo-zigomática, com ressecção parcial da extensão intracraniana do tumor, que invadia e infiltrava a dura-máter. O tumor invadia o seio cavernoso, que foi aberto entre os ramos maxilar e mandibular do trigêmio. Entretanto, por não haver plano entre o tumor e as estruturas do seio, optou-se pela não ressecção desta porção do tumor. Preze (lias após, foi submetido à cirurgia por "degloving" com exérese completa da porção extracraniana do tumor. Vinte dias após, foi submetido à remoção do flap ósseo da craniotomia por apresentar infecção nesta região, não controlada com antibioticoterapia. No 50° PO da cirurgia intracraniana, recebeu alta hospitalar. No 40° PO, já recuperado das cirurgias, iniciou tratamento radioterápico e hormonioterápico. O paciente apresentou dois episódios de. meningite, 12 e 16 meses após a cirurgia, sendo isolados agentes das vias aéreas superiores (Estreptococus pneumoniae e Haemophilus influenza). Cerca de 20 dias após o segundo episódio, foi submetido à correção de desvia de septo nasal e ressecção de sinéquias nasais, que. estavam obstruindo a drenagem adequada das secreções rino-sinusais, levando a sinusites e meningite. O paciente também desenvolveu quadro de otite média secretora, atribuída à manipulação cirúrgica e à radioterapia da nasofaringe, sendo submetido à colocação de tubo de ventilação de demora (em T), bilateral mente. Atualmente, está com quase 5 anos de acompanhamento, não apresentando sinais de recidiva e tendo recuperado a função visual completamente.
CASO 3: F.F.S.S., 16 anos, branco, masculino, procurou nosso Serviço com história de obstrução nasal e epistaxe há 5 meses. Apresentava protusão do olho direito e abaulamento anterior e lateral da região malar direita. A CT e a RNM mostravam extensa massa em fossa nasal direita e rinofaringe, com destruição do corpo e grande asa do esfenóide, invasão do seio esfenoidal e do ápice orbitário direito, com espessamento dos músculos reto medial e reto lateral. Havia, também, acometimento do espaço mastigatório, das fossas ptérigo-palatinas, pterigóides e infratemporal direita. Havia extensão intracraniana do tumor. Foi submetido à angiografia, com oclusão de artéria carótida direita por 25 minutos, não apresentando sinais de isquemia cerebral, e sendo comprovada satisfatória irrigação do parênquima cerebral direito, no controle angiográfico, pela carótida interna esquerda, através da comunicante anterior. Cateterizadas e embolizadas com partículas de Ivalon (300500 microns as artérias maxilar interna, faríngea ascendente e meningea média direitas, com resultados satisfatórios. Não foram embolizados os ramos meníngeos da artéria carótida interna direita. Após 6 dias da embolização, foi submetido a craniotomia órbito-fronto-têmporo-zigomática, notando-se massa tumoral que invadia e ultrapassava a dura-máter, estando em contato com o parênquima cerebral do lobo direito, havendo bom plano de separação. Foi realizada ressecção parcial da extensão infra-craniana do tumor, deixando-se porção junto ao seio cavernoso. A tomografia pós-operatória mostrou tumor infra-craniano residual, envolvendo o seio cavernoso direito, e persistência dos aspectos extra-cranianos anteriores. No 2°PO, desenvolveu hematoma extra-dural, sendo submetido à drenagem, com bom resultado. O paciente desenvolveu diplopia e diminuição da acuidade visual após a cirurgia, com regressão parcial espontânea. Após 45 dias, realizou nova angiografia com embolização dos ramos nutrientes do tumor. Quatro dias após, foi submetido à exérese completa do tumor extra-craniano pela via de Caldwell-Luc ampliada. O tamponamento foi retirado no 3°PO sem intercorrências. CT de controle mostrou tumor residual medindo 2,0 x 2,3 x 2,7 cm localizado lateralmente à porção cavernosa da - artéria carótida direita. O paciente foi, então, encaminhado para tratamento radioterápico e submetido à hormonioterapia. Houve regressão completa do tumor residual ao término do tratamento. Até o momento, após 1 ano do tratamento, não apresenta sinais de recidiva.
DISCUSSÃO
Angiofibroma juvenil é tumor vascular benigno, composto por estroma de tecido conectivo e matriz de estruturas vasculares dilatadas. Esses vasos sanguíneos não apresentam as camadas musculares em sua parede, sendo extremamente frágeis, o que lhe confere grande tendência a hemorragias. Considera-se que o tumor se torne progressivamente mais fibroso, em detrimento do componente vascular e que, usualmente, regrida após o paciente atingir os 20 ou 25 anos de idade, embora esses fatos ainda não estejam provados3, 4, 5. Este tumor apresenta-se recoberto por mucosa na fossa nasal, esfenóide e nasofarínge, enquanto sua extensão lateral é recoberta por forte pseudo-cápsula formada por tecido fibroso6, que se origina pela compressão que exerce sobre os tecidos vizinhos.
Sua etiologia ainda é desconhecida, sendo atribuída à hiperplasia fibroangiomatosa, foco hamartomatoso ectópico ou hiperproliferação de tecido paragangliômico. Sua origem, entretanto, está bem estabelecida. Segundo Neel6, é a parede látero-posterior do teto da fossa nasal, onde o processo esfenoidal do osso palatino encontra a asa horizontal do osso vômer e a raiz do processo pterigóide do osso esfenóide. É a margem superior do forame esfeno-palatino e a região da crista etmoidal (ou inserção da região posterior do corneto médio). Embriologicamente, esse sítio corresponde à inserção da membrana bucofaríngea, que é o limite. entre o ectoderma estomadeo e o endoderma do intestino anterior. Esse sítio também corresponde à junção entre o viscerocrânio membranoso (ossos palatino, vômer e laminas pterigóideas) e o neurocrânio cartilaginoso (osso esfenóide).
O crescimento do angiofibroma também é bens conhecido6. Inicialmente, cresce abaixo da mucosa, na margem posterior da coana, expandindo-se superior e medialmente, até alcançar a borda posterior do septo, formando massa no teto da fossa nasal posterior. Essa massa cresce anteriormente, preenchendo a fossa nasal, deslocando o septo e achatando os cornetos. Cresce inferior e posteriormente para a nasofaringe, deslocando o palato mole e podendo ser visto na orofaringe. No seu crescimento lateral, alarga o forame esfeno-palatino e ocupa a fossa ptérigo-maxilar. Expande-se, deslocando anteriormente a parede posterior do seio maxilar, e destruindo a raiz do processo pterigóideo, posteriormente. A lesão continua a expandir-se lateralmente e, através da fissura pterigomaxilar, alcança a fossa infratemporal, levando ao abaulamento da região da bochecha pode expandir-se superiormente, alcançando a porção inferior da fossa temporal, causando abaulamento acima do zigoma. Nestes tumores extensos, a lesão usualmente cresce superiormente na fossa ptérigo-maxilar e, através da fissura orbitária inferior, alcança a fissura orbital superior, destruindo a grande asa do osso esfenóide e causando proptose, pela invasão da cavidade, orbitária. No crescimento do tumor nas fossas ptérigomaxilar e infratemporal, ocorre destruição do osso que forma a base do processo pterigóide, que corresponde à união do corpo e da grande asa do osso esfenóide. Quando isso ocorre, existe abertura da cavidade craniana, e o tumor cresce, deslocando a dura-máter da fossa média, anteriormente ao forame lacerum e lateralmente ao seio cavernoso e artéria carótida interna. Enquanto isso, o tumor pode crescer diretamente do seu ponto de origem através do assoalho do seio esfenóide, preenchendo-o.
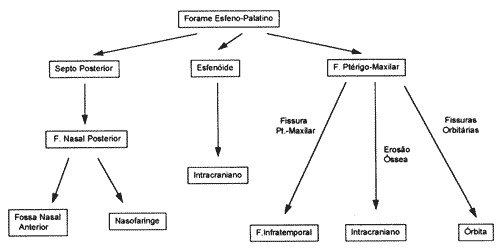
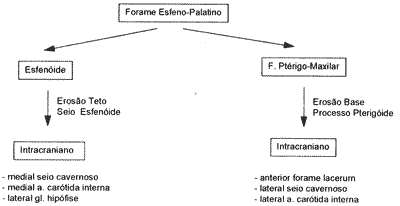
Esquema I: Crescimento do Angiofibroma juvenil
Esquenta II: Vias de invasão intracraniana do Angiofibroma Juvenil
Expande-se, então, superiormente através do teto deste seio, deslocando a glândula hipófise lateralmente, e alcançando a sela túrcica e quiasma óptico (Esquenta I).
Lloyd e Phelps, estudando 44 casos, observaram o acometimento do esfenóide em 73% dos casos, da fossa infratemporal cm 60%, da órbita em 30% e da fossa craniana média em 20%7.
Basicamente, o angiofibroma juvenil pode alcançar a fossa craniana média por duas vias8: através da fossa ptérigomaxilar, anteriormente ao forame lacerum e lateralmente ao seio cavernoso c artéria carótida interna; e/ou através do seio esfenoidal e sela túrcica, medial aro seio cavernoso e artéria carótida interna e lateralmente à glândula hipófise (Esquema II). A fossa craniana anterior também pode ser invadida na evolução deste tumor, embora menos usual9.
O acometimento intracraniando do angiofibroma juvenil é relativamente frequente, sendo relatado na freqüência de 10 a 36% dos casos10-12. Suspeita de invasão intracraniana deve ser feita quando o paciente apresenta menos de 12 anos de idade, extensão lateral do tumor para bochecha ou fossa infratemporal, déficit em função de nervos cranianos e proptose5. Alguns autores acreditam que o tumor é mais agressivo em pacientes mais jovens13, e que a invasão intracraniana seria resultante desta agressividade, que ocorre preferencialmente nesta faixa etária. Outros, entretanto, acreditam que a extensão intracraniana é resultante da proximidade. anatômica e não pelo tumor ser meais agressivo11.
Embora exista invasão intracraniana, o angiofibroma é tumor benigno, fibroso e. de crescimento lento. Exceto na fossa nasal, rinofaringe e seios paranasais, é recoberto por pseudo-cápsula fibrosa espessa, resultante da compressão que exerceu sobre os tecidos adjacentes durante seu crescimento6. A extensão intra-craniana do tumor, que ocorre. pela sua expansão da fossa Ptérigo-maxilar, é recoberta por essa pseudo-cápsula, que desloca a dura-máter, semi invadi-la. Segundo fones, quase todos tumores são extradurais, e podem ser dissecados elas aderências com a dura-máter de maneira segurar através de dissecção romba entre o tumor e a meninge, inclusive do seio cavernos11.
A invasão da dura-máter é pouco usual, e relatos desta ocorrência são bastante raros. Bathia13, em levantamento da literatura até 1967, observou que existiam relato de somente 9 casos de angiofibroma com extensão intracraniana. fones, em 1986, relata que desde a publicação ele Bathia até a sua própria, somente mais 84 casos com extensão intracraniana haviam sido relatados. Destes 84 casos, somente um penetrava a dura-mater, mas não acometia a massa cerebral10, 11.
Em nossa experiência, três casos apresentavam invasão da dura. Dois deles eram recidivas, mostrando que as alterações resultantes da manipulação prévia predispuseram invasão da dura-máter. No.s três casos, o tumor penetrava a dura e podia ser visto em contato direto com o parênquima cerebral, embora não o invadisse e fosse facilmente separado deste. Nos três casos, havia invasão do seio cavernoso e a completa ressecção do tumor deixaria grandes seqüelas. Deste incido, optou-se por ressecção parcial do tumor intracraniano no primeiro tempo, seguida da ressecção completa do tumor extracraniano e posterior tratamento complementar.
Inicialmente, os pacientes foram submetidos à embolização endoarterial seletiva do tumor, que e procedimento de rotina em nosso Serviço há vários anos14. Após cerca de 5 dias, foi submetido ao primeiro tempo cirúrgico, no qual o neurocirurgião ressecou parcialmente o tumor intra-craniano, deixando tini plano bem determinado entre o tumor residual e a massa extra-craniana. A ressecção extra-craniana foi realizada após cerca ele 20 dias elo primeiro ato operatório, para que houvesse adequada cicatrização do enxerto meníngeo e estabilização neurológica. O acesso foi através de rinotomia lateral era um caso e degloving em dois casos. A porção extra-craniana foi removida era sua totalidade nos três casos. Após ó tratamento cirúrgico, foram submetidos à radioterapia externa e hormonionioterapia (anti-andrógeno), por 2 meses.
O tratamento destes tumores é controverso. Alguns autores o consideram irressecáveis5, 15, 16. Outros preconizara a ressecção combinada por via intra e extra-craniana3, 4, 10, 17, 18. Outros, ainda, defendem a ressecção incompleta por via extra-craniana, deixando tumor residual intra-craniano e observando seu comportamento10, 13, 19, 20. Alguns preconizam a associação de modalidades adjuvantes após a ressecção inicial: radioterapia4, 6, 10, 17, hormonioterapia criocirurgia6, 25 quimioterapia26, embolização3, 27 e eletrocoagulação6, 13. O emprego de radioterapia e hormonioterapia pré-operatórios também é preconizado28, e já foi por nós utilizado corno rotina no tratamento dos angiofibromas, atualmente abandonado29. Nossa rotina atual é a ressecção cirúrgica após embolização endoarterial do tumor, o que reduziu drasticamente as perdas sangüíneas30.
A discussão a respeito da ressecção combinada intra e extra-craniana deve basear-se na avaliação da morbidade resultante da craniotomia e a resultante da presença do tumor residual ou recorrente. É importante lembrar os inúmeros relatos de tumores intra-cranianos residuais totalmente assintomáticos11. English20 descreveu 4 casos de ressecção extracraniana incompleta, em que o tumor intracraniano residual apresentou regressão documentada por angiografia. Recentemente, Jacobsson relatou um caso com tumor intracraniano residual extenso, embolizado terapêuticamente com álcool polivinílico por ter sido considerado irressecável, e que apresentou involução documentada com tomografia computadorizada27. Bathia13 observou fibrose do tumor residual que atribuiu à desvascularização resultante da ressecção primária. Neel6 relata que a regressão do tumor residual não é incomum. Inclusive Jafek, que preconiza a ressecção intra e extracraniana combinada, acredita que ressecções subtotais podem ser tratadas de forma espectante10. Conley31 refere que a recorrência é mais desejável do que cirurgia excessivamente perigosa. Jones também defende ressecção extracraniana segura e adequada, com tratamento espectante do tumor residual, que será tratado somente quando os sintomas justificarem os riscos cirúrgicos. A taxa de recorrência no tratamento dos tumores intracranianos é de 50% para Jones, comparada com 50% de Jafek, que preconiza abordagem intra e extracraniana combinada.
Session16 prefere radioterapia para o tratamento de tumores com extensão intra-craniana. Refere que o tumor regrediu completamente ou parcialmente, permanecendo estável em 11 de 12 casos.
Alguns autores preconizam tratamento radioterápico para o tumor inicial e para recidiva do angiofibroma com extensão intracraniana, embora exista risco da indução de tumores, principalmente nesta população de indivíduos jovens. Fitzpatrick relatou controle de 80% de 45 casos tratados com moderada-dose radioterápico (3.000 a 3.500 cGy) e 94%, quando utilizados 2 cursos de radioterpia. Não foi encontrando nenhum caso de indução tumoral em acompanhamento de 2 a 20 anos. Em razão do fato do tumor regredir devido à endarterite obliterante que ocorre até cerca de 24 meses após o término do tratamento, recomendam conduta espectante de observação32.
A presença de receptores hormonais nos angiofibromas está bem estabelecida33, mas a hormonioterapia no seu tratamento é bastante controversa. Schiff34 relata que a utilização de estrógeno, embora não faça o tumor desaparecer, faz com que ele involua, torne-se duro e denso e com menor aporte sangüíneo, fato que pode ser constatado macroscópica e histologicamente. Essas alterações fazem parte do processo de rnaturação do tumor que ocorre na vigência do estrógeno. Essa maturação facilitaria a dissecção e reduziria significativamente as perdas sanguíneas34, 35. Johsen sugere que o tumor é testosterona dependente, pois observou aumento do ritmo de crescimento deste com administração deste hormônio a um paciente, que apresentou involução com a substituição do tratamento por dietilstilbestrol24. Nossos pacientes receberam acetato de ciproterona, que é um esteróide sexual com ação antiandrogênica na dose de 50mg ao dia, durante o período de dois meses.
Em nossa experiência, todos os três casos apresentaram evolução favorável, com regressão do tumor residual e poucas seqüelas definitivas. Esses três jovens pacientes retomaram suas atividades normais e apresentam vida compatível com sua idade, sem restrições determinadas pelo tumor ou seu tratamento.
CONCLUSÃO
Embora a extensão intracraniana do angiofibroma juvenil seja razoavelmente freqüente, a invasão meníngea é muito rara. Esta ocorre principalmente em recidivas tumorais, mas também pode ocorrer na primeira manifestação, sem nenhum tratamento anterior. A invasão do parênquima cerebral não foi descrita, embora seu deslocamento por massas de grande volume possa ocorrer. O plano de descolamento entre o tumor e o parênquima é bem definido. O tratamento destes casos incomuns é controverso, principalmente quando existe acometimento do seio cavernoso. A ressecção parcial do tumor intracraniano, seguida da ressecção completa do tumor extracraniano, e a utilização de tratamentos complementares (radioterapia e hormonioterapia) fornecem controle satisfatório do tumor, com poucas seqüelas.
REFERÊNCIA BIBLIOGRAFIA
1. BATSAKIS, JG - Tumors of the Head and Neck: Clinical and Pathological Considerations, Baltimore, 2nd ed, Willians & Wilkins Co., 1979. pag 291-312.
2. HANDOUSA, A. et al - Nasopharyngeal fibroma: a clinical-patllological study of 70 cases. J Laryngol Otol, 68: 647-666,1961.
3. STANDEFER, J. et al - Combined intracranial and extracranial excision of nasopharyngeal angiofibroma. Laryngoscope, 93: 772-779, 1983.
4. CHANDLER, J. R. et al - Nasopharyngeal angiofibromas: Staging and-management. Ann Otol Rhinol Laryngol, 93: 323-329, 1984.
5. WARD, P. H. - The evolving management of juvenile nasopharyngeal angiofibroma. J Laryngol Otol (Sappl), 8: 103-104, 1983.
6. NEEL, H. B. et al - Juvenile angiofibroma. Review of 120 cases. Am J Surg, 126: 547-556, 1973.
7. L LOYD, G. A. S. and Phelps, P. D. -Juvenile angiofibroma: lnaging by magnetic resonance, CT and conventional techniques. Clin Otolaryngol, 59: 675-683, 1986.
8. CHHANGANI, D. L. et al - Intracranial extensions in nasopharyngeal fibroma. J Laryngol Otol, 82: 11:17, 1968.
9. CLOSE, L. G. et al - Surgical management of nasopharyngeal angiofibroma involving the cavernous sinus. Arch Otolaryngol Head Neck Surg 115: 1091-1095, 1989.
10. JAFEK, B. W. et al - Juvenile nasopharyngeal angiofibroma: management of intracranial extension. Head Neck Surg, 2: 119-128, 1979.
11. JONES, G. C. et al - Juvenile angiofibromas: behavior and treatment of extensive and residual tumors. Arch Otolaryngol Head Neck Surg, 112: 1191-1193, 1986.
12. ECONOMOU, T. S. et al - Juvenile nasopharyngeal angiofibroma: an update of the UCLA experience, 1960-1985. Laryngoscope, 98: 170-175, 1988.
13. BATHIA, M. L. AND MISHRA, S. (:. - Intracranial extensions of juvenile angiofibroma of the nasopharynx. J Laryngol Otol, 81: 1395-1403, 1967.
14. PAIVA, L. J.; BUTUGAN, O. et al - A embolização no tratamento do nasoangiofibroma juvenil. Rev Bras ORL, 42: 30-34, 1976.
15. WALDMAN, S. R. et al - Surgical experience with nasopharyngeal angiofibroma. Arch Otolaryngol Head Neck Surg, 107: 677-682, 1981.
16. SESSIONS, R. B. et al - Radiographic staging of juvenile angiofibroma. Head Neck Surg, 3: 279-283, 1981.
17. STEINBERGER, S. J. and WETMORE, R. F. - Current management of juvenile- nasopharyngeal angiofibroma. Trans Pa Acad Ophamol Otolaryngol, 37: 6570, 1984.
18. KREKORIAN, E. A. and KATO, R. H. - Surgical management of nasopharyngeal angiofibroma with intracranial extension. Laryngoscope, 87: 154-164, 1977.
19. SELLARS, S. L. - Juvenile nasopharyngeal angiofibroma. S Afr Med. J, 58: 961-964, 1980.
20. ENGLISH, G. M. et al - Surgical treatment of invasive angiofibroma. Arch Otolaryngol Head Neck Surg, 96:312-318, 1972.
21. HENDERSON, G. P. Jr. and PATTERSON, G. N. - Further experience in treatment of juvenile nasopharyngeal angiofibroma. Laryngoscope, 79: 561-580, 1969.
22. Johns, M. E. et al - Estrogen receptors in nasopharyngeal angiofibromas. Laryngoscope, 90: 628-634, 1980.
23. WIJ-SHAN - Surgical management of nasopharyngeal angiofibroma. Rhinology, 21: 299-302, 1983.
24. JOHNSEN, S. et al - The action of hormones on juvenile nasopharyngeal angiofibroma. Acta Otolaryngol, 61: 143-159, 1966.
25. SMITH, M. F. W. et al - Gryosurgical techniques in removal of angiofibromas. Laryngoscope, 74: 1071-1080, 1964.
26. GOEPFERT, H. et al - Chemotherapy of locally aggressive head and neck tumors in the pediatric age group: Desmoid fibromatosis and nasopharyngeal angiofibromas. Amn J Surg, 144: 437-444, 1982.
27. JACOBSSON, M. et al - Involution of juvenile nasopharyngeal angiofibroma with intracranial extension. Arch Otolaryngol Head Neck Surg, 115: 238-239, 1989.
28. MARTIN, H. et al - Juvenile nasopharyngeal angiofibroma. Ann Surg, 127:513-536, 1948.
29. PAIVA, L. J.; BUTUGAN, O. et al - Contribuição para o estudo do nasofibroma juvenil. Rev Bras ORL, 40: 390-398, 1974.
30. BUTUGAN, O. - Angiofibroma juvenil - Aspectos atuais terapêuticos. In: Brandão e Ferraz - Cirurgia de Cabeça e Pescoço, São Paulo, 1a ed, Livraria Roca Ltda, 1989. pg 417-420.
31. CONLEY, J. et al - Nasopharyngeal angiofibroma in the juvenile. Surg Gynecol Obstet, 126: 825-837, 1968.
32. FITZPATRICK, P. J. et al - The nasopharyngeal angiofibroma. Arch Otolaryngol, 106: 234-236, 1980.
33. BRENTANI, M. M.; BUTUGAN, O, et al - Multiple steroid receptors in nasopharyngeal angiofibromas. Laryngoscope, 99: 398-401, 1989.
34. SCHIFF, M. - Juvenile nasopharyngeal angiofibroma. A theory of pathogenesis. Laryngoscope, 69: 981-1016, 1959.
35. GRANATO, L. - Angiofibroma juvenil - Vias de abordagem, técnicas e complicações. In Brandão e Ferraz, Cirurgia de Cabeça e Pescoço, São Paulo, 1a ed, Livraria Roca Ltda, 1989. pg 397-414, 1989.
* Professor Associado da Disciplina de otorrinolaringologia da Faculdade de Medicina da U.S.P.
** Pós-Graduando e Médico da Divisão de Clínica otorrinolaringológica do Hosp. Clínicas - F.M.U.S.P.
*** Professor Doutor da Disciplina de Otorrinolaringologia da Faculdade de Medicina da U.S.P.
**** Professor Titular da Disciplina de otorrinolaringologia da Faculdade de Medicina da U.S.P.
Trabalho realizado no Departamento de Otorrinolaringologia do Hospital das Clínicas da Faculdade de Medicina da Universidade de São Paulo e LIM 32 (Serviço do Prof Aroldo Miniti)
Av. Dr. Enéas de Carvalho Aguiar, 255 - 6 andar - sala 60(12 -Saca Paulo - SP - CEP: 03020-000
Artigo recebido em 30 de março de 1995.
Artigo aceito em 01 de maio de 1995.
<
Imprimir: ![]()