

Ano: 1988 Vol. 54 Ed. 4 - Outubro - Dezembro - (2º)
Seção: Artigos Originais
Páginas: 109 a 111
ALTERAÇÕES OTONEUROLÓGICAS EM DEFICIÊNCIA SISTÊMICA DE CARNITINA: RELATO DE UM CASO*
Otoneurologic alteration in carnitine deficiency: report of a case*
Autor(es):
Yotaka Fukuda**
Pedro Henrique de Miranda Mota***
![]()
Introdução
A carnitina é uma amônia quaternária cuja importância fisiológica reside em ser cofator no transporte de ácidos graxos livres de cadeia longa do citoplasma para o interior da mitocôndria. Dentro da mitocôndria, estes ácidos graxos de cadeia longa serão oxidados e utilizados como fonte de energia no ciclo de Krebs.
A deficiência de carnitina é reconhecida clinicamente pelos seguintes sintomas: fraqueza muscular progressiva, episódios recurrentes de acidose metabólica e encefalopatia, seguidos de estupor e coma (DiMauro 1985).
O estudo otoneurológico de uma paciente, com deficiência de carnitina foi suscitado pelo quadro clínico sugestivo de alterações do sistema vestibular: tonturas, vômitos e alterações da marcha. Foram encontradas algumas alterações, e o presente artigo destina-se a documentá-las e a tecer alguns comentários sobre correlações existentes entre as deficiências de carnitina e o aparelho cócleo-vestibular.
Caso clínico
1 -Identificação: G.A.O., 5 anos, sexo feminino, cor branca, natural e procedente de São Paulo-SP.
2 -Anamnese:
2.1 - Queixa principal: vômitos.
2.2 - História Pregressa da Moléstia Atual: refere o pai que há cerca de um ano a criança vem apresentando quadro de vômitos em jato, incoercíveis, associado a dor abdominal e febre, com duração de 4 a 5 dias. Nestes episódios a criança era levada ao hospital para hidratação. Em março de 1987, a paciente passou a apresentar a sintomatologia acima descrita, associada à alteração da marcha e a quedas freqüentes. Após 24 a 48 horas do início do quadro apresentou episódio de convulsão que durou cerca de 30 segundos, a seguir ficando sonolenta e confusa. Com o quadro acima descrito foi solicitada sua internação no Hospital São Paulo.
2.3 - Antecedentes pessoais: a mãe teve gestação sem intercorrências. O parto foi normal, gemelar, a termo, sem sofrimento fetal. Intoxicação por "diabo verde" (produto químico de limpeza) aos 2 anos, sendo tratada no Hospital das Clínicas-USP. Alimentação normoprotéica e normocalórica. Quadro de vacinação completo.
2.4 - Antecedentes familiares: irmã gêmea com os mesmos sintomas e falecida em decorrência de vômitos, aos 5 anos de idade. As duas tiveram início do quadro na mesma época.
3 - Exame Físico: paciente no momento da internação com regular estado geral, desidratada, apática, afebril e corada.
Pele e fâneros: sem alterações. Gânglios: micropoliadenia cervical indolores e móveis.
Músculos,, ossos e articulações: fraqueza muscular.
Coração e pulmões: normais. Abdome: fígado palpável a 2 centímetros abaixo da reborda costal direita.
Exame ORL: normal.
Exame neurológico: força muscular presente e diminuida. Marcha com alargamento da base e leve desequilíbrio.
Foram feitas as seguintes hipóteses diagnosticas: distúrbio metabólico e processo expansivo cerebral.
Com este quadro foi solicitado estudo otoneurológico.
4 - Estudos laboratoriais gerais: Estudo do líquido céfalo-raquidiano: líquor límpido e incolor. Células: 04 Hémacias: 00 Glicose: mg/100 ml Cloretos: 731 Sódio: 1 mEq/1 Pesquisa da cisticercose: negativa.
Reação de Wassermann: negativa
Reação de Pandy: negativa
Reação de Nonne: negativa
Reação de Weichbrodt: negativa
Mucoproteinas séricas: 2,1 mg/ 100 ml.
Antiestreptolisina 0: 20 unidades TODD.
Hemograma:
hemácias 3,5 milhões/mm3
Hb 10,2 g% Ht 30 ml de eritrócitos/100 ml de sangue Valor globular 0,97 CHCM 34% HCM 29 picogramas VCM 85 microcúbicos.
Anisocitose e hìpocromia presentes.
Leucograma: 32001eucócitos/mm3
Neutrófilos bastões 05
Neutrófilos segmentados 38
Eosinófilos 01
Basófilos 00
Linfócitos 55
Monócitos 01
Velocidade de hemossedimentação: 5 mm em uma hora.
Glicemia: 91 mg/dl
Triglicérides: 152 mg/dl
Colesterol total: 279 mg/dl
HDL colesterol: 44 mg/dl
LDL colesterol: 201 mg/dl
ECG: arritmia sinusal
EEG: normal em sonolência e sono espontâneo, sem fotoestimulação.
Biópsia muscular: presença de depósitos de gordura nas fibras musculares estriadas.
5 - Diagnóstico: hipótese diagnóstica de lipidose muscular, sugestivo de deficiência de carnitina.
6 - Tratamento: foi administrada carnitina por via oral, com remissão dos sintomas.
7 - Evolução: Em setembro de 1987 a medicação foi suspensa por três dias, com reaparecimento dos vômitos. Foi reinstituída a medicação, com desaparecimento dos sintomas.
8 - Exame Otontológico
8.1 - Exame audiológico: Audiometria tonal liminar (Audiômetro ISO-1964) MA18
Discriminação de palavras:
OD (monossílabo) 88% a 90 dB
OE (monossílabo) 92% a 80 dB
Limiar de recepção de palavras (SRT)
OD 50 dB
OE 40 dB
Impedanciometria: Timpanometria normal.
Volume equivalente do ouvido médio ÷ 0,5 ml no OD e 0,9 ml no OE.
Reflexos estapédicos:
2.8.2 Exame vestibular:
Calibração: regular
Nistagmo espontâneo ausente com olhos abertos e presente horizontal para esquerda com a velocidade angular da componente lenta igual a 8 graus por segundo.
Nistagmo semi-espontâneo: ausente.
Nistagmo optocinético: simétrico com VACL igual a 7 graus por segundo nos sentidos horário e anti-horário.
Rastreio pendular: curva tipo 1/11 no sentido horizontal e curva tipo II no sentido vertical.
Nistagmo de posição: ausente.
Nistagmo pós-calórico: na irrigação com água aquecida a 44 graus centígrados observado nistagmo com VACL igual a 8 graus por segundo à esquerda e 9 graus por segundo à direita. Na irrigação do ouvido com água a 33 graus centígrados observado nistagmo com VACL igual a 10 graus por segundo à esquerda e 7 graus por segundo à direita.
Conclusão: Síndrome vestibular periférica irritativa.
Audiometria de Tronco Cerebral (ABR): normal.
Discussão
A carnitina (beta-hidroxi-gama-Ntrimetilamônia butirato) é uma amônia quaternária sintetizada a partir da lisina e metionina derivadas da degradação das proteínas. As fontes de carnitina são as originadas da dieta normal (25%) e as sintetizadas no fígado e nos rins (75%); é excretada principalmente pelos rins (Hanh 1985; Werneck 1985; Winter 1987).
A carnitina possui múltiplas funções ligadas à lipólise, termogênese, cetogênese, e provavelmente ao metabolismo do nitrogênio (Lancet 1982; Borum 1985). Os carboidratos e lipídios são importantes fontes energéticas. Durante exercícios físicos, os primeiros são utilizados inicialmente. Quando estão esgotadas as reservas de carboidratos, o organismo utiliza os lipídios plasmáticos, que são os ácidos graxos e triglicérides. Estes lipídios são transportados para o interior das mitocôndrias pela ação da carnitina-palmitil-transferase I e II, sofrendo a seguir beta-oxidação, sendo utilizados no ciclo de Krebs (Werneck 1981).
Se ocorrer deficiência de carnitina, as gotículas de gordura localizadas no citoplasma das fibras musculares não serão mobilizadas, e surgirão sintomas de fraqueza muscular. Além da fraqueza muscular, outros sintomas são encontrados em pacientes portadores de deficiência de carnitina: encefalopatia com hipoglicemia semelhante à da síndrome de Reye, aumento dos níveis de amônia plasmática e de TGO e TGP. Os vômitos e a cefaléia podem estar relacionados com a lactiacidemia transitória (Werneck 1985). Esta acidose lática é decorrente do impedimento da utilização do piruvato no ciclo de Krebs, e pela crescente utilização de glicogênio e da atividade glicolítica no músculo com metabolismo aeróbio defeituoso (DiMauro 1985).
São reconhecidas 2 entidades clínicas de deficiência de carnitina: a forma sistêmica e a forma miopática. Na forma miopática encontram-se baixos níveis de carnitina nas fibras musculares e níveis normais no plasma. Na forma sistêmica há comprometimento do fígado, coração, rins, músculos esqueléticos, SNC e outras estruturas com níveis de carnitina baixos no plasma (Cruse 1984; DiDonato 1984; DiMauro 1985; Werneck 1985).
Existe uma forma primária de deficiência de carnitina, com evidência de herança autossômica recessiva (Almog 1979; DiDonato 1982; Cruse 1984). A forma secundária tem etiologia variada: síndrome de Fanconi, dieta parenteral prolongada ou dieta baseada no leite de soja unicamente, fatores hepatotóxicos que inibem a síntese de carnitina como o ácido valpróico, usado como droga anticonvulsivante, absorção intestinal deficiente de carnitina, diarréia crônica severa (Lancet 1982; Borum 1985; Winter 1987). Em neonatos contribuem o plasma materno deficiente em carnitina e a prematuridade (Winter 1987). A deficiência de carnitina também costuma aparecer em pacientes submetidos à hemodiálise prolongada.
O tratamento foi inicialmente baseado em corticoterapia, em que houve redução da gordura acumulada nas fibras musculares e melhora clínica evidente (Harrinson 1980; Werneck 1981). A reposição de carnitina na forma levógira mostrou remissão dos sintomas em alguns casos (Winter 1987).
Conclusão
As disfunções mitocondriais podem ser devidas à deficiência de carnitina, uma enzima necessária no transporte de ácidos graxos de cadeia longa do citoplasma para o interior das mitocôndrias, onde serão metabolizados para fornecimento de energia. Pacientes portadores de deficiência de carnitina geralmente apresentam quadro de vômitos intensos, fraqueza muscular progressiva, encefalopatia e acidose metabólica. Podem também apresentar disacusia neuro-sensorial.
No presente estudo apresentamos o caso de uma criança de 5 anos de idade, do sexo feminino, cuja irmã gêmea faleceu em decorrência de vômitos incoercíveis. A paciente, além do quadro de vômitos incoercíveis, desequilíbrio, desidratação e fraqueza muscular progressiva, apresentou síndrome vestibular periférica irritativa e disacusia neuro-sensorial moderada com boa discriminação de palavras.
Na orelha interna, a estria vascular é uma estrutura altamente vascularizada e com grande concentração de mitocôndrias, necessária para manutenção do potássio na endolinfa. Quando houver insuficiente aporte glicidico, a gordura é mobilizada-para suprimento energético. Pacientes portadores de deficiência de carnitina poderão ser afetados nesta situação, podendo ocasionar disfunções labirínticas.
Summary
Mitochondrial disorders can be caused by carnitine deficiency. Carnitine is a cofactor in the transport of long-chain free fatty acids into mitochondria, where they will be metabolized in order to produce energy for the cell. Patients with carnitine deficiency generally present episodes of intense vomiting, progressive muscular weakness, encephalopathy and metabolic acidosis.
We report a case of a 5-year-old patient, female, whose twin sister died of recurrent episodes of intense vomiting. The patient presented intense vomiting, loss of balance, dehydratation and progressive muscular weakness. She also presented vertigo (irritative vestibular syndrome) and a moderate sensorineural hearing loss with a good vocal discrimination.
In the inner ear, the stria vascularis is a highly vascularized structure with a great mitochondrial concentration, and it is necessary to keep a high level of potassium in the endolymph. When there is not a sufftcient supply of glucose, the fatty acid are used in order to produce energy. This is how patients with carnitine deficiency can eventually present labyrinthine disorders.
Bibliografia
1. ALMOG, C.; FRIED, K.; REIF, R.; ZIEGHELBOLM, J.; LEWINSOHN, G. Autossomal recessive lipid storage myopathy (probable carnitine deficiency). Journal of Medical Genetics 16:435-438,1979.
2. BORUM, A. Role of carnitine during development. Can J Physiol Pharmaco1 63: 571-576,1985.
3. CRUSE, R.P.; DIMAURO, S.; TOWFIGHI, J.; TREVISAN, C. Familial Systemic Carnitine Deficiency. Arch Neurol 41: 301305,1984.
4. DI DONATO, S.; RIMOLDI, M.; CORNELIO, F.; BOTTACCHI, E.; GIUNTA, A. Evidence for autossomal recessive inheritance in systemic carnitine deficiency. Ann Neurol 11: 190-192,1982.
5. DI DONATO, S.; PELUCCHETTI, D.; RIMOLDI, M.; MORA, M.; GARAVAGLIA, B.; FINOCCHIARO, G. Systemic carnitine deficiency: clinical and biochemical and morphological cure with L-carnitine. Neurology 34: 157-162, 1984.
6. DI MAURO, S.; BONILLA, E.; ZEVIANE, M.; NAKAGAWA, M.; DE VIVO, D.C. Mitochondrial myopathies. Ann Neurol17: 521-538, 1985.
7. HANH, P. Carnitine and ketones in perinatal periods. Fed Proc 44: 2369-2373, 1985. 8. LANCET Carnitine: clue or cure? (Editorial) Nov 6:1027-28,1982.
9. WERNECK, L.C.; BOER, C.A.A.; PAPADIMITRIOU, A.; Dl MAURO, S.; Miopatia por deficiência de Carnitine-palmitil-transferase. Arq. Neuropsiquiatria, Vol 41, n° 5:377-385, 1981.
10. WERNECK, L.C.; DI MAURO, S. Deficiência muscular de carnitina. Arq. Neuropsiquiatria, Vo143, n° 3:281-295,1985.
11. WINTER, S.C.; SZABO-ACZEL, S.; CURRY, C.J.R.; HUTCHINSON, H.T.; HAZUE, R.; SHUNG, A. Plasma carnitine deficiency. Am. J. Dis. Child.141: 660-665, 1987.
12. HARRINSON. Principies of Internai Medicine. Ninth Edition. McGraw-Hill Book Company. Pages 2064-65 and 1760-61, 1980.
* Trabalho realizado na disciplina de Otorrinolaringologia da Escola Paulista de Medicina (Chefe do serviço: Prof. Dr. Pedro Luiz Mangabeira Albernaz)
** Professor Adjunto da Disciplina de Otorrinolaringologia da Escola Paulista de Medicina.
*** Residente do 2° ano da Disciplina de Otorrinolaringologia da Escola Paulista de Medicina.
ALTERAÇÕES OTONEUROLÓGICAS EM DEFICIÊNCIA SISTÊMICA DE CARNITINA: RELATO DE UM CASO*
Otoneurologic alteration in carnitine deficiency: report of a case*
Yotaka Fukuda**
Pedro Henrique de Miranda Mota***
Introdução
A carnitina é uma amônia quaternária cuja importância fisiológica reside em ser cofator no transporte de ácidos graxos livres de cadeia longa do citoplasma para o interior da mitocôndria. Dentro da mitocôndria, estes ácidos graxos de cadeia longa serão oxidados e utilizados como fonte de energia no ciclo de Krebs.
A deficiência de carnitina é reconhecida clinicamente pelos seguintes sintomas: fraqueza muscular progressiva, episódios recurrentes de acidose metabólica e encefalopatia, seguidos de estupor e coma (DiMauro 1985).
O estudo otoneurológico de uma paciente, com deficiência de carnitina foi suscitado pelo quadro clínico sugestivo de alterações do sistema vestibular: tonturas, vômitos e alterações da marcha. Foram encontradas algumas alterações, e o presente artigo destina-se a documentá-las e a tecer alguns comentários sobre correlações existentes entre as deficiências de carnitina e o aparelho cócleo-vestibular.
Caso clínico
1 -Identificação: G.A.O., 5 anos, sexo feminino, cor branca, natural e procedente de São Paulo-SP.
2 -Anamnese:
2.1 - Queixa principal: vômitos.
2.2 - História Pregressa da Moléstia Atual: refere o pai que há cerca de um ano a criança vem apresentando quadro de vômitos em jato, incoercíveis, associado a dor abdominal e febre, com duração de 4 a 5 dias. Nestes episódios a criança era levada ao hospital para hidratação. Em março de 1987, a paciente passou a apresentar a sintomatologia acima descrita, associada à alteração da marcha e a quedas freqüentes. Após 24 a 48 horas do início do quadro apresentou episódio de convulsão que durou cerca de 30 segundos, a seguir ficando sonolenta e confusa. Com o quadro acima descrito foi solicitada sua internação no Hospital São Paulo.
2.3 - Antecedentes pessoais: a mãe teve gestação sem intercorrências. O parto foi normal, gemelar, a termo, sem sofrimento fetal. Intoxicação por "diabo verde" (produto químico de limpeza) aos 2 anos, sendo tratada no Hospital das Clínicas-USP. Alimentação normoprotéica e normocalórica. Quadro de vacinação completo.
2.4 - Antecedentes familiares: irmã gêmea com os mesmos sintomas e falecida em decorrência de vômitos, aos 5 anos de idade. As duas tiveram início do quadro na mesma época.
3 - Exame Físico: paciente no momento da internação com regular estado geral, desidratada, apática, afebril e corada.
Pele e fâneros: sem alterações. Gânglios: micropoliadenia cervical indolores e móveis.
Músculos,, ossos e articulações: fraqueza muscular.
Coração e pulmões: normais. Abdome: fígado palpável a 2 centímetros abaixo da reborda costal direita.
Exame ORL: normal.
Exame neurológico: força muscular presente e diminuida. Marcha com alargamento da base e leve desequilíbrio.
Foram feitas as seguintes hipóteses diagnosticas: distúrbio metabólico e processo expansivo cerebral.
Com este quadro foi solicitado estudo otoneurológico.
4 - Estudos laboratoriais gerais: Estudo do líquido céfalo-raquidiano: líquor límpido e incolor. Células: 04 Hémacias: 00 Glicose: mg/100 ml Cloretos: 731 Sódio: 1 mEq/1 Pesquisa da cisticercose: negativa.
Reação de Wassermann: negativa
Reação de Pandy: negativa
Reação de Nonne: negativa
Reação de Weichbrodt: negativa
Mucoproteinas séricas: 2,1 mg/ 100 ml.
Antiestreptolisina 0: 20 unidades TODD.
Hemograma:
hemácias 3,5 milhões/mm3
Hb 10,2 g% Ht 30 ml de eritrócitos/100 ml de sangue Valor globular 0,97 CHCM 34% HCM 29 picogramas VCM 85 microcúbicos.
Anisocitose e hìpocromia presentes.
Leucograma: 32001eucócitos/mm3
Neutrófilos bastões 05
Neutrófilos segmentados 38
Eosinófilos 01
Basófilos 00
Linfócitos 55
Monócitos 01
Velocidade de hemossedimentação: 5 mm em uma hora.
Glicemia: 91 mg/dl
Triglicérides: 152 mg/dl
Colesterol total: 279 mg/dl
HDL colesterol: 44 mg/dl
LDL colesterol: 201 mg/dl
ECG: arritmia sinusal
EEG: normal em sonolência e sono espontâneo, sem fotoestimulação.
Biópsia muscular: presença de depósitos de gordura nas fibras musculares estriadas.
5 - Diagnóstico: hipótese diagnóstica de lipidose muscular, sugestivo de deficiência de carnitina.
6 - Tratamento: foi administrada carnitina por via oral, com remissão dos sintomas.
7 - Evolução: Em setembro de 1987 a medicação foi suspensa por três dias, com reaparecimento dos vômitos. Foi reinstituída a medicação, com desaparecimento dos sintomas.
8 - Exame Otontológico
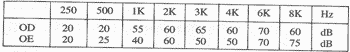
8.1 - Exame audiológico: Audiometria tonal liminar (Audiômetro ISO-1964) MA18
Discriminação de palavras:
OD (monossílabo) 88% a 90 dB
OE (monossílabo) 92% a 80 dB
Limiar de recepção de palavras (SRT)
OD 50 dB
OE 40 dB
Impedanciometria: Timpanometria normal.
Volume equivalente do ouvido médio ÷ 0,5 ml no OD e 0,9 ml no OE.
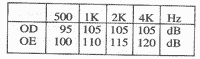
Reflexos estapédicos:
(TABELA 2, PÁG. 110)
2.8.2 Exame vestibular:
Calibração: regular
Nistagmo espontâneo ausente com olhos abertos e presente horizontal para esquerda com a velocidade angular da componente lenta igual a 8 graus por segundo.
Nistagmo semi-espontâneo: ausente.
Nistagmo optocinético: simétrico com VACL igual a 7 graus por segundo nos sentidos horário e anti-horário.
Rastreio pendular: curva tipo 1/11 no sentido horizontal e curva tipo II no sentido vertical.
Nistagmo de posição: ausente.
Nistagmo pós-calórico: na irrigação com água aquecida a 44 graus centígrados observado nistagmo com VACL igual a 8 graus por segundo à esquerda e 9 graus por segundo à direita. Na irrigação do ouvido com água a 33 graus centígrados observado nistagmo com VACL igual a 10 graus por segundo à esquerda e 7 graus por segundo à direita.
Conclusão: Síndrome vestibular periférica irritativa.
Audiometria de Tronco Cerebral (ABR): normal.
Discussão
A carnitina (beta-hidroxi-gama-Ntrimetilamônia butirato) é uma amônia quaternária sintetizada a partir da lisina e metionina derivadas da degradação das proteínas. As fontes de carnitina são as originadas da dieta normal (25%) e as sintetizadas no fígado e nos rins (75%); é excretada principalmente pelos rins (Hanh 1985; Werneck 1985; Winter 1987).
A carnitina possui múltiplas funções ligadas à lipólise, termogênese, cetogênese, e provavelmente ao metabolismo do nitrogênio (Lancet 1982; Borum 1985). Os carboidratos e lipídios são importantes fontes energéticas. Durante exercícios físicos, os primeiros são utilizados inicialmente. Quando estão esgotadas as reservas de carboidratos, o organismo utiliza os lipídios plasmáticos, que são os ácidos graxos e triglicérides. Estes lipídios são transportados para o interior das mitocôndrias pela ação da carnitina-palmitil-transferase I e II, sofrendo a seguir beta-oxidação, sendo utilizados no ciclo de Krebs (Werneck 1981).
Se ocorrer deficiência de carnitina, as gotículas de gordura localizadas no citoplasma das fibras musculares não serão mobilizadas, e surgirão sintomas de fraqueza muscular. Além da fraqueza muscular, outros sintomas são encontrados em pacientes portadores de deficiência de carnitina: encefalopatia com hipoglicemia semelhante à da síndrome de Reye, aumento dos níveis de amônia plasmática e de TGO e TGP. Os vômitos e a cefaléia podem estar relacionados com a lactiacidemia transitória (Werneck 1985). Esta acidose lática é decorrente do impedimento da utilização do piruvato no ciclo de Krebs, e pela crescente utilização de glicogênio e da atividade glicolítica no músculo com metabolismo aeróbio defeituoso (DiMauro 1985).
São reconhecidas 2 entidades clínicas de deficiência de carnitina: a forma sistêmica e a forma miopática. Na forma miopática encontram-se baixos níveis de carnitina nas fibras musculares e níveis normais no plasma. Na forma sistêmica há comprometimento do fígado, coração, rins, músculos esqueléticos, SNC e outras estruturas com níveis de carnitina baixos no plasma (Cruse 1984; DiDonato 1984; DiMauro 1985; Werneck 1985).
Existe uma forma primária de deficiência de carnitina, com evidência de herança autossômica recessiva (Almog 1979; DiDonato 1982; Cruse 1984). A forma secundária tem etiologia variada: síndrome de Fanconi, dieta parenteral prolongada ou dieta baseada no leite de soja unicamente, fatores hepatotóxicos que inibem a síntese de carnitina como o ácido valpróico, usado como droga anticonvulsivante, absorção intestinal deficiente de carnitina, diarréia crônica severa (Lancet 1982; Borum 1985; Winter 1987). Em neonatos contribuem o plasma materno deficiente em carnitina e a prematuridade (Winter 1987). A deficiência de carnitina também costuma aparecer em pacientes submetidos à hemodiálise prolongada.
O tratamento foi inicialmente baseado em corticoterapia, em que houve redução da gordura acumulada nas fibras musculares e melhora clínica evidente (Harrinson 1980; Werneck 1981). A reposição de carnitina na forma levógira mostrou remissão dos sintomas em alguns casos (Winter 1987).
Conclusão
As disfunções mitocondriais podem ser devidas à deficiência de carnitina, uma enzima necessária no transporte de ácidos graxos de cadeia longa do citoplasma para o interior das mitocôndrias, onde serão metabolizados para fornecimento de energia. Pacientes portadores de deficiência de carnitina geralmente apresentam quadro de vômitos intensos, fraqueza muscular progressiva, encefalopatia e acidose metabólica. Podem também apresentar disacusia neuro-sensorial.
No presente estudo apresentamos o caso de uma criança de 5 anos de idade, do sexo feminino, cuja irmã gêmea faleceu em decorrência de vômitos incoercíveis. A paciente, além do quadro de vômitos incoercíveis, desequilíbrio, desidratação e fraqueza muscular progressiva, apresentou síndrome vestibular periférica irritativa e disacusia neuro-sensorial moderada com boa discriminação de palavras.
Na orelha interna, a estria vascular é uma estrutura altamente vascularizada e com grande concentração de mitocôndrias, necessária para manutenção do potássio na endolinfa. Quando houver insuficiente aporte glicidico, a gordura é mobilizada-para suprimento energético. Pacientes portadores de deficiência de carnitina poderão ser afetados nesta situação, podendo ocasionar disfunções labirínticas.
Summary
Mitochondrial disorders can be caused by carnitine deficiency. Carnitine is a cofactor in the transport of long-chain free fatty acids into mitochondria, where they will be metabolized in order to produce energy for the cell. Patients with carnitine deficiency generally present episodes of intense vomiting, progressive muscular weakness, encephalopathy and metabolic acidosis.
We report a case of a 5-year-old patient, female, whose twin sister died of recurrent episodes of intense vomiting. The patient presented intense vomiting, loss of balance, dehydratation and progressive muscular weakness. She also presented vertigo (irritative vestibular syndrome) and a moderate sensorineural hearing loss with a good vocal discrimination.
In the inner ear, the stria vascularis is a highly vascularized structure with a great mitochondrial concentration, and it is necessary to keep a high level of potassium in the endolymph. When there is not a sufftcient supply of glucose, the fatty acid are used in order to produce energy. This is how patients with carnitine deficiency can eventually present labyrinthine disorders.
Bibliografia
1. ALMOG, C.; FRIED, K.; REIF, R.; ZIEGHELBOLM, J.; LEWINSOHN, G. Autossomal recessive lipid storage myopathy (probable carnitine deficiency). Journal of Medical Genetics 16:435-438,1979.
2. BORUM, A. Role of carnitine during development. Can J Physiol Pharmaco1 63: 571-576,1985.
3. CRUSE, R.P.; DIMAURO, S.; TOWFIGHI, J.; TREVISAN, C. Familial Systemic Carnitine Deficiency. Arch Neurol 41: 301305,1984.
4. DI DONATO, S.; RIMOLDI, M.; CORNELIO, F.; BOTTACCHI, E.; GIUNTA, A. Evidence for autossomal recessive inheritance in systemic carnitine deficiency. Ann Neurol 11: 190-192,1982.
5. DI DONATO, S.; PELUCCHETTI, D.; RIMOLDI, M.; MORA, M.; GARAVAGLIA, B.; FINOCCHIARO, G. Systemic carnitine deficiency: clinical and biochemical and morphological cure with L-carnitine. Neurology 34: 157-162, 1984.
6. DI MAURO, S.; BONILLA, E.; ZEVIANE, M.; NAKAGAWA, M.; DE VIVO, D.C. Mitochondrial myopathies. Ann Neurol17: 521-538, 1985.
7. HANH, P. Carnitine and ketones in perinatal periods. Fed Proc 44: 2369-2373, 1985. 8. LANCET Carnitine: clue or cure? (Editorial) Nov 6:1027-28,1982.
9. WERNECK, L.C.; BOER, C.A.A.; PAPADIMITRIOU, A.; Dl MAURO, S.; Miopatia por deficiência de Carnitine-palmitil-transferase. Arq. Neuropsiquiatria, Vol 41, n° 5:377-385, 1981.
10. WERNECK, L.C.; DI MAURO, S. Deficiência muscular de carnitina. Arq. Neuropsiquiatria, Vo143, n° 3:281-295,1985.
11. WINTER, S.C.; SZABO-ACZEL, S.; CURRY, C.J.R.; HUTCHINSON, H.T.; HAZUE, R.; SHUNG, A. Plasma carnitine deficiency. Am. J. Dis. Child.141: 660-665, 1987.
12. HARRINSON. Principies of Internai Medicine. Ninth Edition. McGraw-Hill Book Company. Pages 2064-65 and 1760-61, 1980.
* Trabalho realizado na disciplina de Otorrinolaringologia da Escola Paulista de Medicina (Chefe do serviço: Prof. Dr. Pedro Luiz Mangabeira Albernaz)
** Professor Adjunto da Disciplina de Otorrinolaringologia da Escola Paulista de Medicina.
*** Residente do 2° ano da Disciplina de Otorrinolaringologia da Escola Paulista de Medicina.
Imprimir: ![]()
All rights reserved - 1933 /
2026
© - Associação Brasileira de Otorrinolaringologia e Cirurgia Cérvico Facial